
IF:15.4《Cell Death Differ》浦东医院朱敏敏:中药活性成分(苦杏仁苷/甘草酸)自组装水凝胶治疗创伤性脑损伤
专栏:学术前沿
发布日期:2025-10-17
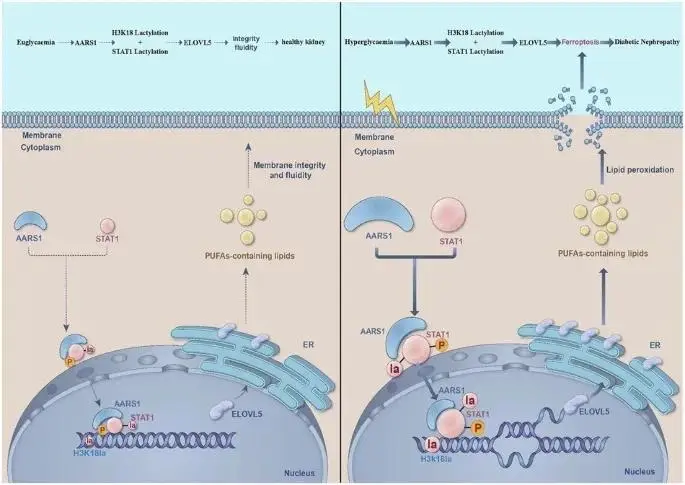
糖尿病肾病(DN)是全球终末期肾病的首要病因。近年发现,乳酸介导的组蛋白乳酸化作为一种新型表观遗传修饰参与糖尿病并发症,但催化该修饰的“乳酰转移酶”在DN中仍属空白;本研究聚焦新发现的乳酰转移酶AARS1,探讨其是否通过组蛋白H3第18位赖氨酸乳酸化(H3K18la)驱动DN发生。

复旦大学附属浦东医院朱敏敏教授团队利用野生型与AARS1杂合缺失(AARS1+/–)小鼠构建DN模型,结合转录组、脂质组及分子生物学手段,揭示AARS1通过两条途径激活肾细胞铁死亡:①AARS1作为乳酰转移酶,直接催化H3K18la及STAT1乳酸化,促进脂肪酸延长酶ELOVL5的转录,导致脂质过氧化;②AARS1与STAT1形成复合物协同增强ELOVL5表达。抑制AARS1(基因减半或β-丙氨酸干预)或阻断STAT1(氟达拉滨)均可减轻铁死亡、延缓DN进展,从而为DN提供“乳酰化-铁死亡”新靶点。该文章于2025年9月23日以《ARS1-mediated lactylation of H3K18 and STAT1 promotes ferroptosis in diabetic nephropathy》为题发表于《Cell Death Differ》(DOI: 10.1038/s41418-025-01587-4)。
Scheme 1 外科辅助水凝胶(AG-gel)通过清创术后局部给药抑制异常补体激活以增强TBI后神经保护的示意图
本研究首次证实丙氨酰-tRNA合成酶1(AARS1)是一种乳酰转移酶,在DN肾组织中高表达;通过构建AARS1杂合缺失(AARS1+/–)DN小鼠并结合转录组、脂质组及分子生物学手段,发现AARS1催化H3K18la及非组蛋白STAT1乳酸化,协同STAT1上调ELOVL5转录,促进PUFA脂质过氧化与铁死亡,导致肾功能障碍和细胞死亡。抑制AARS1(基因减半或β-丙氨酸)或阻断STAT1(氟达拉滨)均可减轻铁死亡、延缓DN进展,从而为DN提供“乳酰化-铁死亡”新靶点与干预策略。
(1) 在DN患者和模型中,AARS1和H3K18la的表达均升高
DN患者及模型肾组织AARS1、总乳酸化及H3K18la水平随病程升高(图1A–D)。HE与Masson染色显示胶原沉积和纤维化随分期递增,TUNEL示肾小管与肾小球细胞死亡同步增加(图1A)。高糖刺激使HGEC与HK-2细胞AARS1、乳酸化、H3K18la及死亡均升高,甘露醇对照无影响。

图1 AARS1-乳酸化-H3K18la轴在DN肾组织中随病程升高。 A 人肾穿连续切片(HE、Masson、TUNEL)及AARS1、pan-Kla、H3K18la IHC示DN分期愈高则结构损伤、纤维化与细胞死亡愈重,乳酸化信号同步增强(bar=50 μm);B 模型小鼠肾同源染色复现上述变化;C Western blot与D qPCR证实DN鼠肾AARS1蛋白、pan-Kla、H3K18la及mRNA均显著上调(*P<0.05–****P<0.0001)
(2)AARS1的上调参与了DN的发生
AARS1+/- DN小鼠肾组织损伤、纤维化及细胞死亡减轻,AARS1、乳酸化与H3K18la水平下降,肾功能指标改善(图2A–C)。高糖条件下敲低AARS1或给予抑制剂Gln-AMS,均同步降低AARS1与H3K18la并减少HGEC和HK-2细胞死亡(图2D–F)。
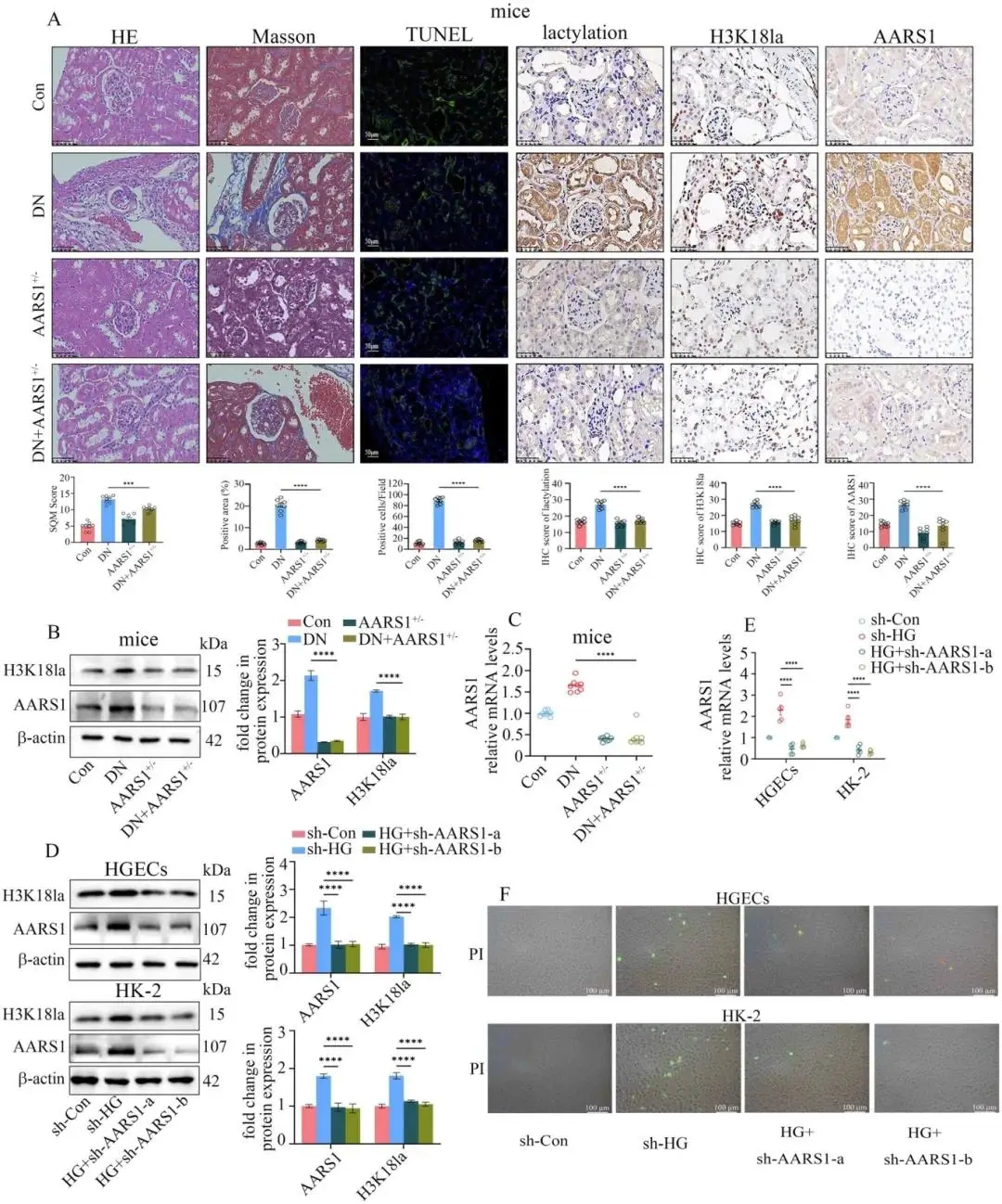
图2 AARS1杂合缺失减轻DN肾损伤与乳酰化。(a)HE、Masson、TUNEL及IHC示AARS1+/–DN组结构损伤、纤维化和细胞死亡减少,AARS1、乳酰化、H3K18la同步下调(标尺50 μm);(b)Western blot示肾组织AARS1、H3K18la蛋白降低;(c)qPCR示AARS1 mRNA减少;(d)高糖升高AARS1与H3K18la,被si-AARS1逆转;(e)高糖诱导AARS1 mRNA上升,敲低后恢复;(f)PI染色示高糖致细胞死亡增加,AARS1沉默抑制(标尺100 μm)
(3)AARS1上调通过引发铁下垂参与DN的发病机制
RNA-seq显示DN肾组织富集不饱和脂肪酸合成、延长及脂肪酸代谢通路(图3A)。随DN分期进展,4-HNE、MDA、ACSL4升高,GPX4降低(图3B)。Fer-1缓解DN小鼠肾损伤,抑制ACSL4并恢复GPX4,减轻线粒体皱缩、MDA、LPO沉积及Fe²⁺积聚,改善肾功能;在高糖细胞中,Fer-1浓度-时间依赖性减少死亡、脂质过氧化、MMP崩溃、MDA及Fe²⁺与线粒体超氧阴离子。AARS1+/- DN小鼠肾内ACSL4下调、GPX4上调,TEM示线粒体损伤减轻,MDA、LPO、Fe²⁺含量下降(图3C-G)。敲减AARS1或Gln-AMS逆转高糖诱导的 ferroptosis 标志变化。AARS1-H3K18la轴通过触发铁死亡参与DN。
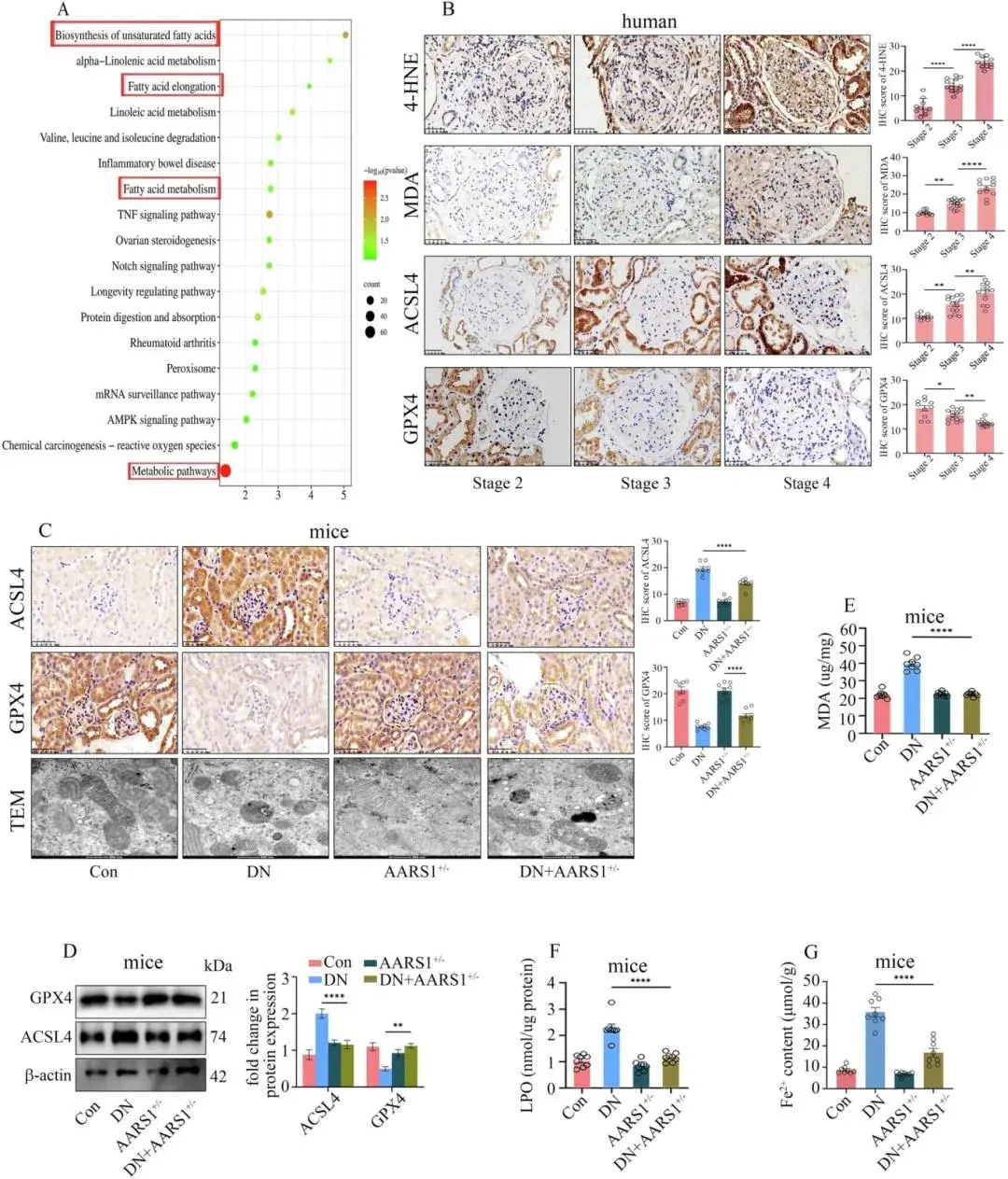
图3 AARS1缺失抑制DN铁死亡。(a)KEGG差异基因分子功能;(b)DN患者肾活检4-HNE、MDA、ACSL4递增,GPX4递减(50 μm);(c)AARS1+/– DN小鼠较DN组ACSL4降、GPX4升,线粒体嵴与体积恢复(IHC 50 μm,TEM 500 nm);(d)Western blot示肾组织ACSL4下调、GPX4上调;(e–g)MDA、LPO、Fe²⁺含量均降低
(4)AARS1 诱导的 H3K18la 通过调节 ELOVL5 转录参与糖尿病肾病模型中的铁死亡
AARS1催化H3K18乳酸化后,选择性占据ELOVL5启动子P3区并上调其转录。ELOVL5表达随DN分期升高,定位内质网,通过增加PUFA(如花生四烯酸)合成驱动肾小管上皮细胞铁死亡;敲低ELOVL5或补充AA可逆转这一进程。AARS1过表达依赖ELOVL5增强铁死亡,抑制AARS1则降低ELOVL5水平并减轻DN模型肾损伤。

图4 AARS1-ELOVL5轴调控铁死亡机制。(a)Western blot示AARS1过表达升高H3K18la、ELOVL5、ACSL4并降低GPX4,si-ELOVL5逆转该变化;(b)qPCR验证AARS1与ELOVL5转录水平;(c)C11-BODIPY 581/591探针显示AARS1过表达加剧脂质过氧化,ELOVL5敲低抑制该效应;(d)JC-1探针示AARS1过表达致线粒体膜电位下降,si-ELOVL5恢复红色聚合物荧光;(e)MDA水平随AARS1过表达升高,被ELOVL5沉默逆转;(f)FeRhoNox-1探针检测Fe²⁺含量,AARS1组荧光增强,si-ELOVL5减弱信号;(g)MitoSOX示AARS1过表达增加线粒体超氧化物,ELOVL5敲低降低红色荧光;(h)PI染色显示AARS1过表达促进细胞死亡,ELOVL5沉默减少死亡;标尺均为100 μm
(5)AARS1在体内和体外均与信号换能器和转录激活因子1(STAT1)直接相互作用
质谱鉴定STAT1为AARS1互作转录因子,Co-IP、免疫荧光和GST pull-down在HGECs与HK-2细胞中验证其直接结合;DN患者肾活检、模型小鼠肾及高糖细胞中STAT1蛋白随疾病分期升高,结果见于图5A–J。
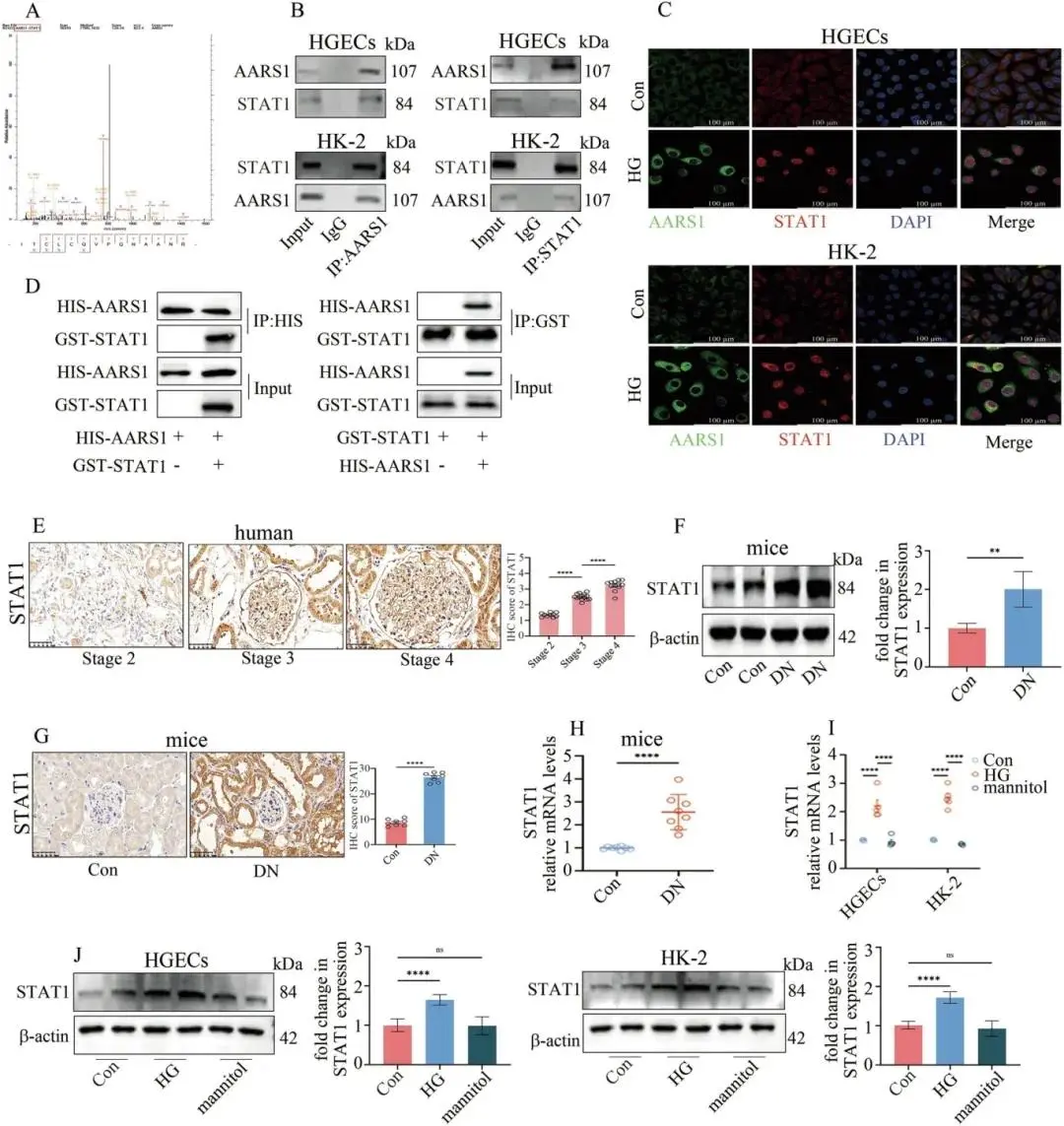
图5 STAT1与AARS1互作及表达上调。(a)质谱筛查潜在结合;(b)Co-IP验证二者相互作用;(c)免疫荧光共定位(标尺100 μm);(d)GST pull-down确认直接结合;(e)DN患者肾活检STAT1随分期递增(标尺50 μm);(f)Western blot示DN小鼠肾STAT1蛋白升高;(g)IHC验证DN小鼠肾STAT1上调(标尺50 μm);(h)qPCR示DN小鼠肾STAT1 mRNA增加;(i)高糖细胞STAT1 mRNA上升;(j)高糖细胞STAT1蛋白上升
(6)在DN模型中,STAT1通过调节ELOVL5的转录参与铁下垂
STAT1直接结合ELOVL5启动子区两处位点并激活其转录,敲低STAT1下调ELOVL5并抑制高糖HGEC/HK-2细胞铁死亡,过表达STAT1则相反且该效应可被si-ELOVL5阻断;Re-ChIP显示STAT1与AARS1共占位。STAT1抑制剂fludarabine呈浓度-时间依赖性降低STAT1及ELOVL5表达,阻断细胞铁死亡;DN小鼠腹腔注射fludarabine同步下调肾内STAT1/ELOVL5,减轻铁死亡、肾损伤并改善肾功能。
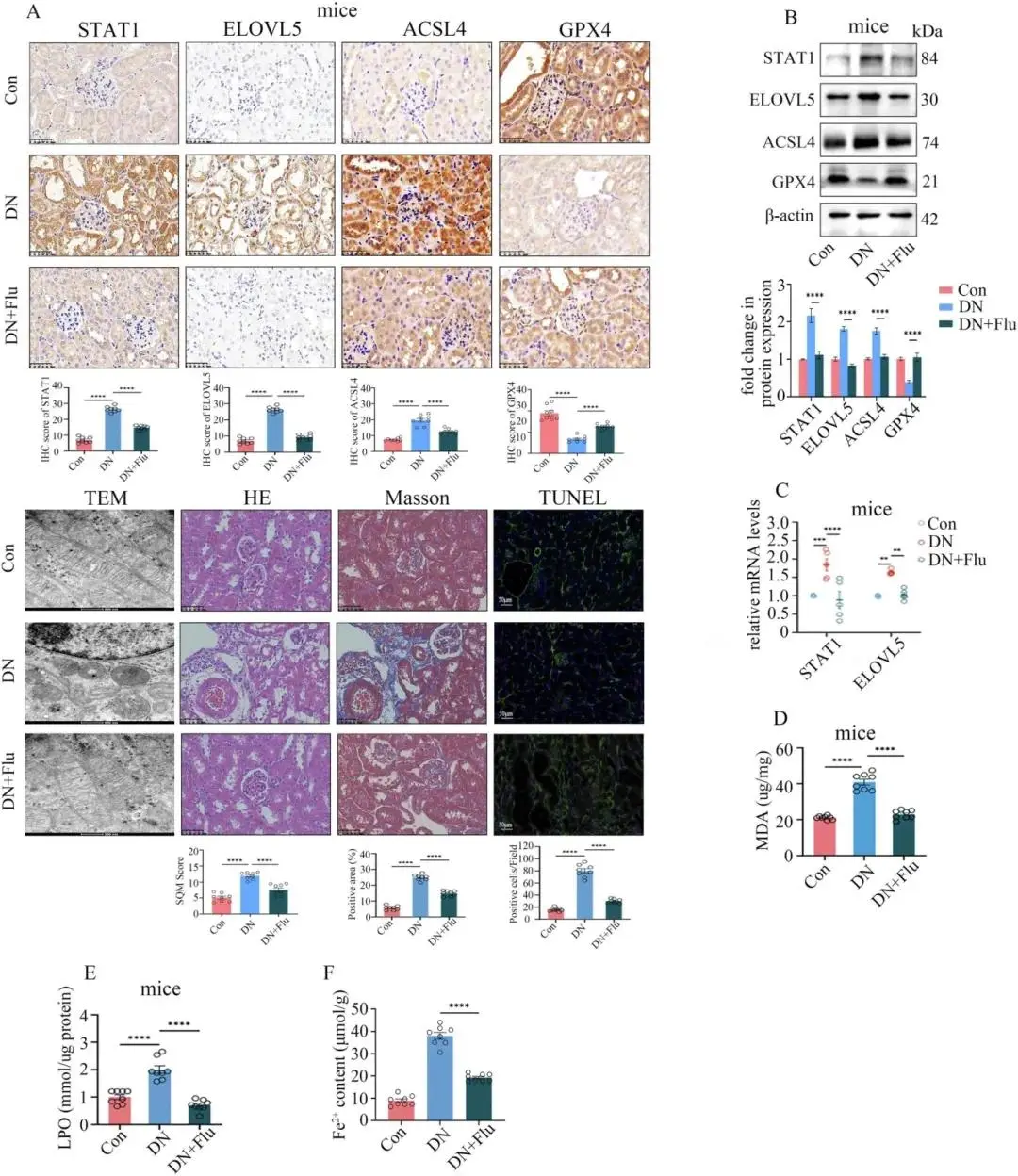
图6 STAT1抑制剂缓解DN肾损伤与铁死亡。(a)HE、Masson、TUNEL、IHC及TEM示DN+Flu组肾损伤、纤维化与细胞死亡减轻,STAT1、ELOVL5、ACSL4下调,GPX4回升,线粒体嵴与体积恢复(HE等标尺50 μm,TEM 500 nm);(b)Western blot示DN+Flu肾组织STAT1、ELOVL5、ACSL4降低,GPX4升高;(c)qPCR示STAT1与ELOVL5 mRNA同步下调;(d–f)DN+Flu组肾MDA、LPO及Fe²⁺含量均显著减少
(7) AARS1调节STAT1和H3K18的乳酸化,从而调节ELOVL5的转录并触发铁凋亡
进一步通过检测血脑屏障相关指标、外源性及内源性C3表达水平,验证了AG-gel能够显著抑制C3表达。通过分子对接、SPR技术更进一步证实了AG-gel与C3直接结合能力。
AARS1依赖其乳酰转移酶活性催化STAT1-K193位点乳酰化,促进STAT1磷酸化及核转位,并增强STAT1与ELOVL5启动子结合;酶失活突变体AARS15M丧失上述功能。体外重组实验显示AARS1以乳酸浓度依赖方式实现STAT1乳酰化,高糖细胞内过表达AARS1同步升高STAT1乳酰化、H3K18la及ELOVL5水平并驱动铁死亡,而突变STAT1-K193阻断这些效应。相关数据见图7A–。
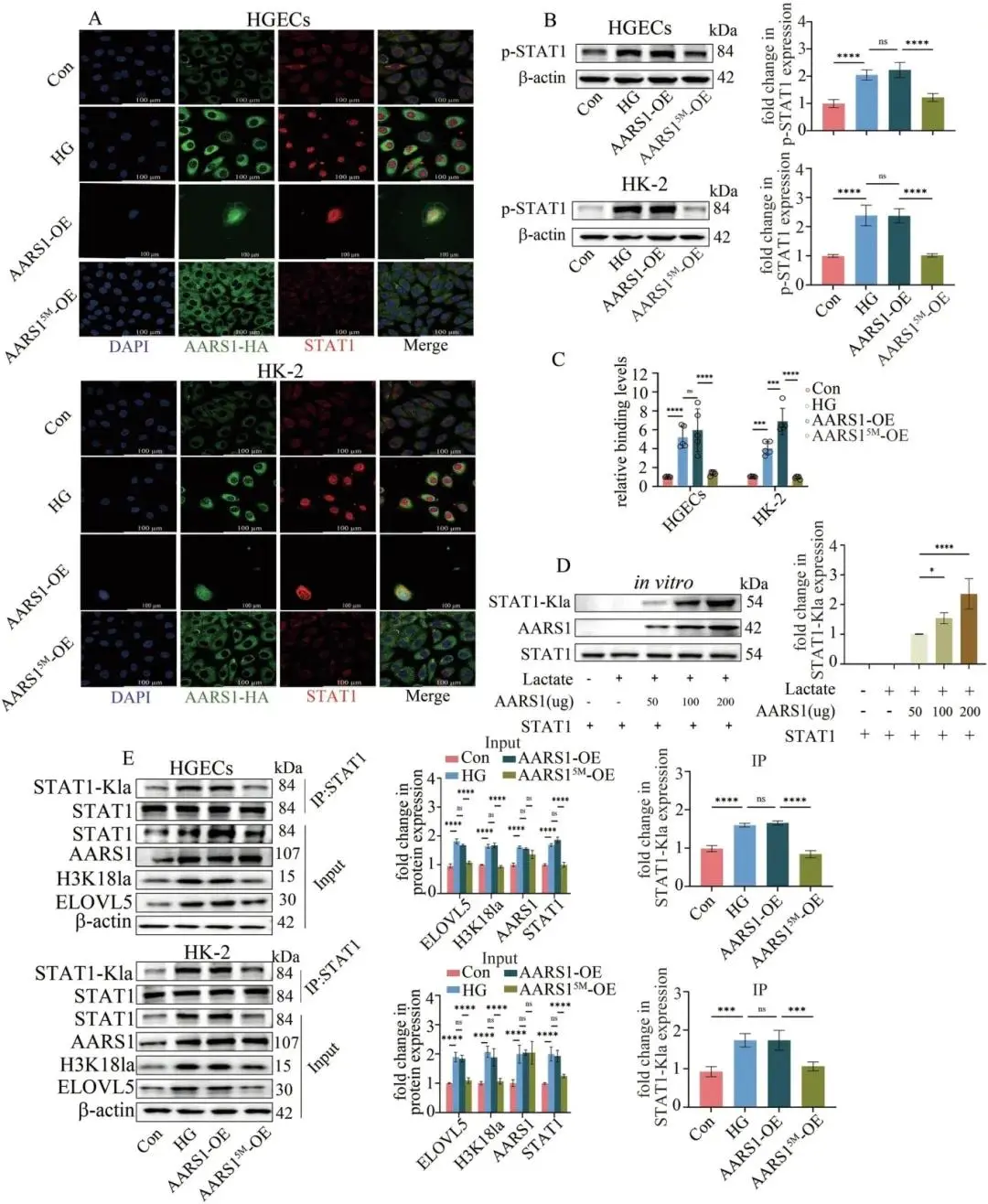
图7 AARS1乳酰化STAT1增强其转录活性。(a)免疫荧光示AARS1过表达驱动STAT1核转位,AARS15M无效(标尺100 μm);(b)Western blot示AARS1提升STAT1磷酸化,AARS15M无此作用;(c)ChIP‒qPCR示AARS1增强STAT1与ELOVL5启动子结合,AARS15M无此效应;(d)体外重组证实AARS1以乳酸依赖方式乳酰化STAT1;(e)Co-IP示AARS1诱导细胞内STAT1乳酰化并上调STAT1、H3K18la及ELOVL5,AARS15M均无效
(8)β-丙氨酸通过抑制aars1诱导的乳酸化,减轻DN模型小鼠和高血糖细胞的铁下垂
丙氨酸与乳酸竞争结合AARS1,呈浓度-时间依赖性抑制高糖细胞内AARS1介导的STAT1与H3K18乳酰化,下调AARS1-STAT1-H3K18la-ELOVL5轴并阻断铁死亡;DN小鼠给予β-丙氨酸后肾组织同步降低上述蛋白表达,减轻铁死亡与肾损伤,肾功能改善。

图8 β-丙氨酸抑制AARS1乳酰化减轻DN肾损伤。(a)HE、Masson、TUNEL、IHC及TEM示DN+β-丙氨酸组肾损伤、纤维化与细胞死亡减少,AARS1、H3K18la、STAT1、ELOVL5、ACSL4下调,GPX4回升,线粒体嵴与体积增加(HE等标尺50 μm,TEM 500 nm);(b)Western blot示肾组织AARS1、H3K18la、STAT1、ELOVL5、ACSL4降低,GPX4升高;(c)qPCR示AARS1、STAT1、ELOVL5 mRNA下调;(d–f)肾内MDA、LPO及Fe²⁺含量均减少
AARS1通过乳酰转移酶活性催化H3K18la与STAT1-K193乳酰化,协同增强STAT1转录活性,上调ELOVL5表达并增加花生四烯酸等PUFA合成,从而驱动肾小球内皮细胞与近端小管上皮细胞铁死亡,加速糖尿病肾病进展。DN患者及高糖模型中AARS1、H3K18la、STAT1、ELOVL5表达随病程递增,β-丙氨酸竞争性抑制AARS1活性可全面阻断该轴,减轻铁死亡与肾损伤并改善肾功能,提示靶向抑制AARS1乳酰化具有治疗DN的临床潜力。

|
创赛生物 提供高品质的医疗产品和服务 |
联系我们 |
产品中心 |
扫码关注
关注公众号 扫码加客服
|

