
IF:14.1《AS》桂林医科大学汪丽燕:双金属纳米酶放大器用于协同铁死亡-铜死亡和代谢重编程以重塑免疫抑制肿瘤微环境
专栏:学术前沿
发布日期:2025-10-23
肝细胞癌(HCC)是典型的“冷肿瘤”,对免疫治疗反应低下,根源在于其高度免疫抑制的微环境(TME)。一方面,肿瘤相关巨噬细胞(TAM)向促瘤的 M2 表型极化,而抑瘤的 M1 表型减少;另一方面,葡萄糖匮乏、乳酸富集的 TME 使 CD4⁺/CD8⁺ T 细胞无法维持有氧糖酵解,效应功能受损,而调节性 T(Treg)细胞却能在此环境中稳定发挥免疫抑制作用。仅清除乳酸虽可逆转部分免疫抑制,但现有 CD8⁺ T 细胞数量不足,且肿瘤对单一免疫原性细胞死亡(ICD)模式存在耐药,难以产生持久抗肿瘤免疫。

针对上述问题,汪丽燕教授团队构建 DSPE-PEG₂₀₀₀-GA 修饰的 Au/Cu/ZIF-8 纳米药物 PEG@AuCZ@CC,通过“代谢重编程+多模式细胞死亡”协同策略破解 HCC 免疫耐受。① CHCA 阻断乳酸外排,造成胞内乳酸堆积并负反馈抑制糖酵解,降低肿瘤葡萄糖消耗,形成“高葡萄糖-低乳酸”TME,促使 M2-TAM 向 M1 复极化,恢复 CD8⁺ T 细胞功能并 destabilize Treg。② 纳米酶 Au 仿葡萄糖氧化酶(GOx)持续耗糖产 H₂O₂,与 Cu²⁺诱导的铜死亡、CO 触发的铁死亡协同,高效释放 ATP、CRT、HMGB1 等 DAMPs,强力激活 ICD,招募并扩增肿瘤内 CD4⁺/CD8⁺ T 细胞及记忆 T 细胞。上述代谢-免疫双重调控将“冷” HCC 转为“热”肿瘤,建立持久系统免疫监视,显著抑制肿瘤进展(图1)。相关研究在2025年10月15日以“Bimetallic Nanozyme Amplifier for Synergistic Ferroptosis-Cuproptosis and Metabolic Reprogramming to Reshape Immunosuppressive Tumor Microenvironment”为题发表于《Advanced science》(DOI: 10.1002/advs.202512764)上。
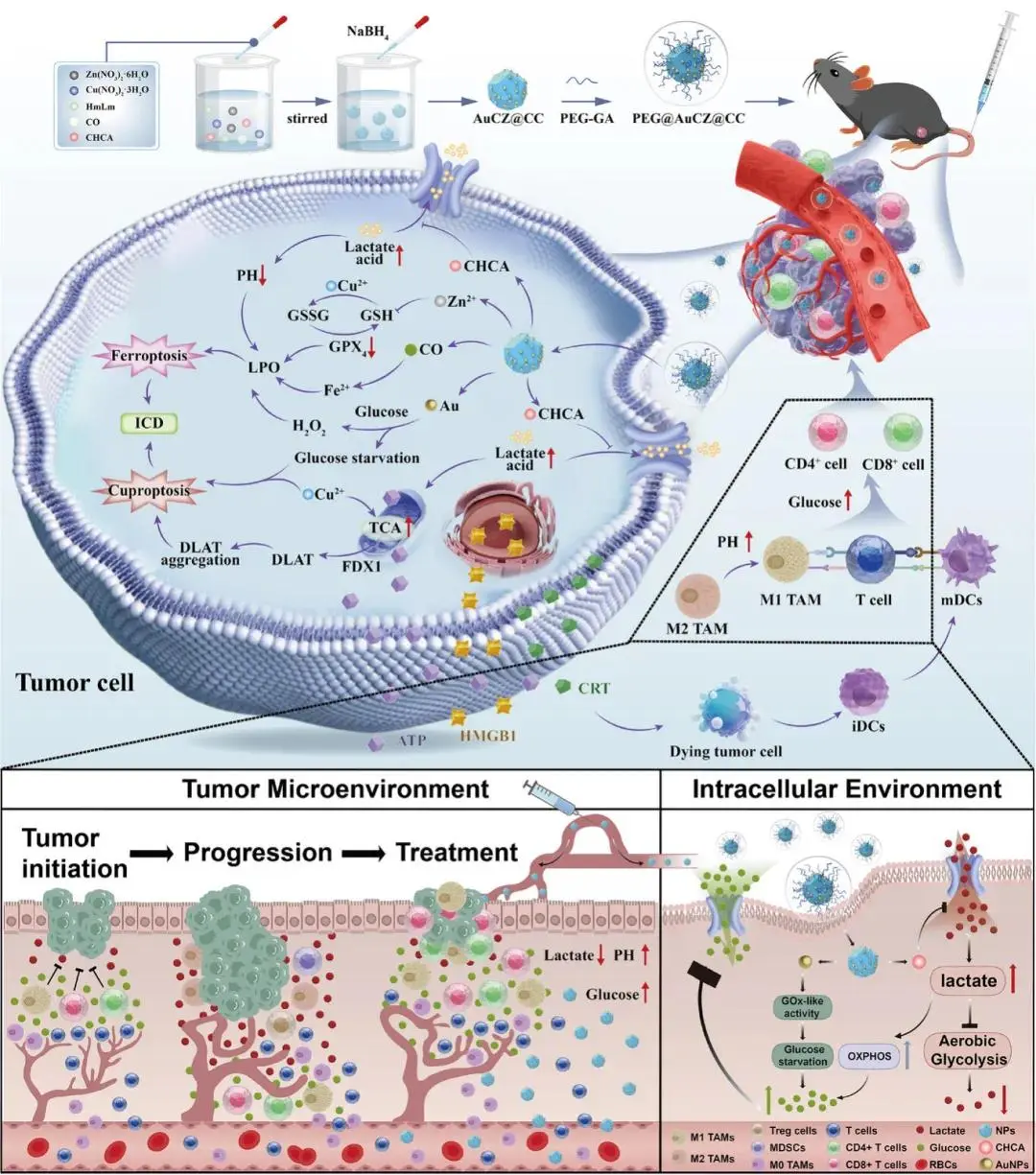
图1 PEG@AuCZ@CC的合成程序和设计与作用原理揭示了如何协同结合铁死亡和杯死亡来增强免疫原性细胞死亡(ICD),同时改善肿瘤免疫抑制微环境,从而共同增强抗肿瘤免疫反应的整体图谱
(1)PEG@AuCZ@CC NPs的合成与表征
研究人员采用水热法合成ZIF-8(图2A),其典型菱面十二面体表面光滑,粒径约135 nm(图2B、E)。随后通过一锅法共掺Cu²⁺并原位还原HAuCl₄,在ZIF-8表面均匀沉积2–3 nm Au纳米颗粒,得到AuCZ@CC;产物保持ZIF-8晶型,出现Cu、Au新信号,水合直径增至242 nm(图2C、F、K)。继续负载CO/CHCA并以DSPE-PEG2000-GA包覆后,形成311 nm的PEG@AuCZ@CC,表面覆有半透明PEG-GA膜,电位由+4.66 mV降至–8.34 mV,FTIR出现PEG特征峰(2872、1114 cm⁻¹),元素映射显示Zn、Cu、Au共存且分布均匀,证实壳层完整(图2D、G、H、I、J)。UV-vis在≈200 nm与≈300 nm处检出CO与CHCA特征吸收,包覆后峰强度减弱;标准曲线测得载药率CO 4.7 %、CHCA 6.6 %,包封率51 %与70 %(图2O)。该纳米粒在纯水及10 % FBS-DMEM中72 h内粒径、PDI、电位几乎不变,5天血清稳定性良好(图2M、N)。透析实验显示,pH 5.4下Zn²⁺/Cu²⁺快速释放、结构崩解,pH 7.4下保持稳定,具备酸触发释药能力(图2P、Q)。
(2)PEG@AuCZ@CC NPs的酶样活性
研究人员首先通过XPS表征了PEG@AuCZ@CC中金属元素的价态与比例(图2R-U)。Zn 2p谱图出现结合能差为23.1 eV的双峰,确认Zn以Zn²⁺形式存在(图1S);Cu 2p谱中931.97/951.80 eV和933.36/953.50 eV信号分别对应Cu⁰/⁺与Cu²⁺,表明Cu的多价共存为后续耗竭谷胱甘肽(GSH)并触发铜死亡奠定基础(图2T);Au 4f₇/₂(83.60 eV)和4f₅/₂(87.27 eV)均指向零价Au,且Au 4f₇/₂相较标准值负移0.4 eV,提示Au纳米颗粒已与ZIF-8表面发生电子相互作用,成功锚定(图2U)。
随后,研究人员系统评估了该材料的葡萄糖氧化酶(GOx)样活性(图2V-X)。在密闭体系中,PEG@AuCZ@CC在1 h内消耗约40 %葡萄糖,溶解氧显著下降,体系pH降低,并在560 nm处出现H₂O₂特征吸收峰,而ZIF-8与PEG@CZ@CC无此性能,证实Au⁰纳米酶活性(图2V、W、X)。GSH耗竭实验进一步显示,随着PEG@AuCZ@CC浓度升高,412 nm处DTNB特征吸收逐渐减弱甚至消失,定量结果与紫外-可见光谱一致,直观颜色变化亦验证Cu²⁺/Cu⁺介导的GSH清除能力(图2Y)。综上,研究人员证实PEG@AuCZ@CC兼具GOx样产H₂O₂与GSH耗竭双重功能,为后续强化Fenton反应提供了先决条件(图2Z)。
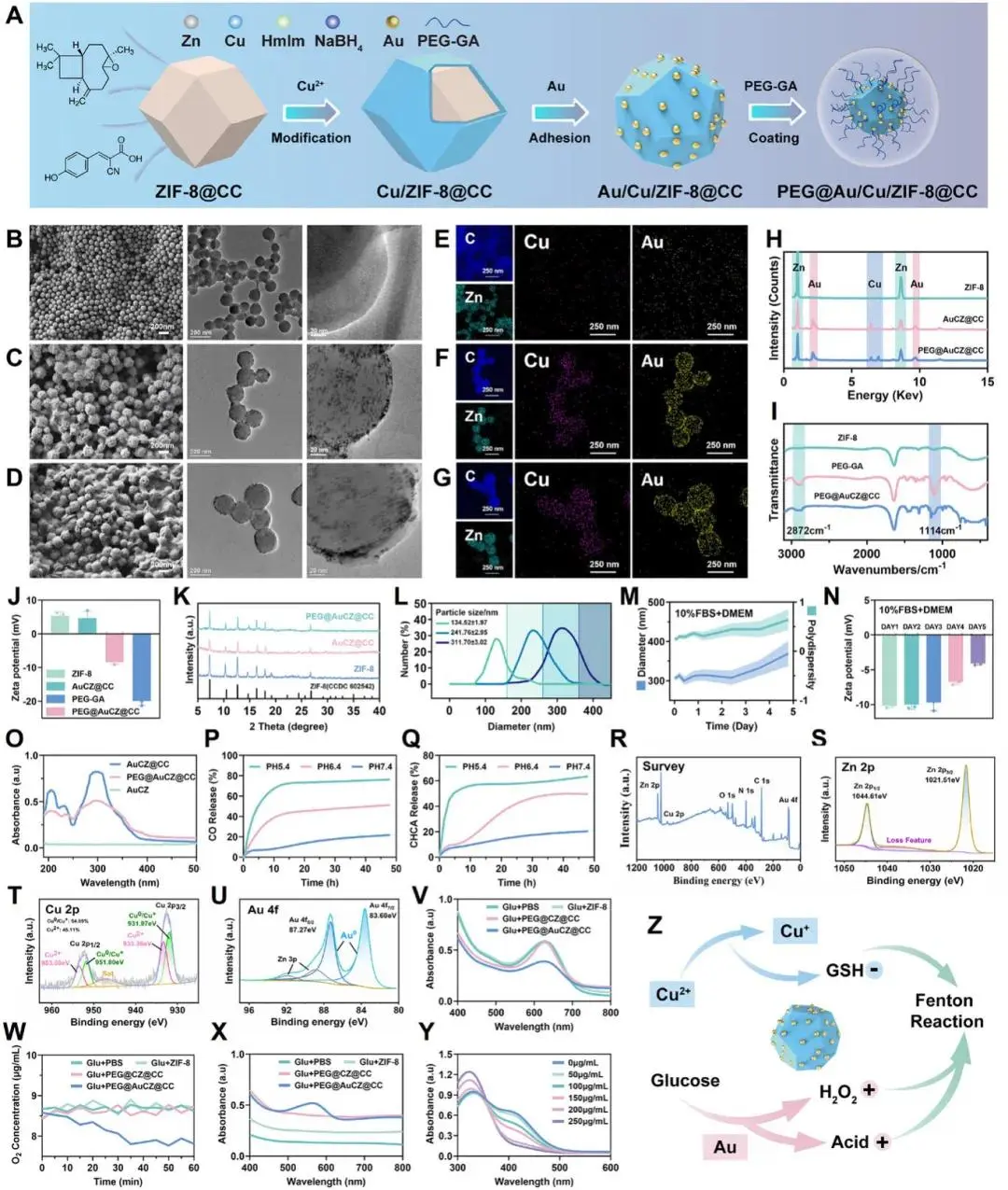
图2 PEG@AuCZ@CC NPs 的物理化学性质表征。A) PEG@AuCZ@CC 制备示意图。B) ZIF-8、C) AuCZ@CC 和 D) PEG@AuCZ@CC 的代表性 SEM 图像和 TEM 图像。E) ZIF-8、F) AuCZ@CC 和 G) PEG@AuCZ@CC 的元素映射。H) 纳米药物的能量衍射光谱 (EDS)。I) ZIF-8、PEG-GA 和 PEG@AuCZ@CC NPs 的傅里叶变换红外光谱。J) 纳米药物和 DSPE-PEG2000-GA 的 Zeta 电位。 (K)ZIF-8、AuCZ@CC 和 PEG@AuCZ@CC NPs 的 XRD 谱。L)通过动态光散射 (DLS) 测量 ZIF-8、AuCZ@CC 和 PEG@AuCZ@CC 在 PBS 中的水合粒径。PEG@AuCZ@CC 在添加 10% FBS 的 DMEM 中培养 5 天后的 M) 稳定性和 N) zeta 电位。O) AuCZ、AuCZ@CC 和 PEG@AuCZ@CC 的紫外可见吸收光谱。在不同时间和不同 pH 下,PEG@AuCZ@CC 对 P) CO 和 Q) CHCA 的累积释放曲线。(R)PEG@AuCZ@CC NPs 的 XPS 全光谱和 S) Zn 2p、T) Cu 2p 和 U) Au 4f 的高分辨率 XPS 光谱。 V) PEG@AuCZ@CC NPs在葡萄糖溶液中葡萄糖消耗测试。W) 葡萄糖与ZIF-8、PEG@CZ@CC和PEG@AuCZ@CC共孵育过程中氧浓度随时间变化的曲线。(X) 不同处理条件下的H2O2生成能力。Y) 不同浓度GSH和PEG@AuCZ@CC NPs存在下DTNB的紫外可见光谱。Z) 模拟酶的生物催化过程示意图
(3)PEG@AuCZ@CC NPs的细胞摄取及抗肿瘤机制
研究人员先在 Hepa1-6 细胞中验证摄取:罗丹明 B 标记显示 PEG@AuCZ@CC 的胞内荧光随时间增强(图 3A);流式定量 15 min 即见显著信号。Zn²⁺、Cu²⁺及 Fe²⁺探针均证实离子浓度显著升高,提示载体降解并释放载荷。正常肝细胞(AML-12、THLE-2)60 % 以上在 120 µg mL⁻¹ 仍存活,而 Hepa1-6 的 IC₅₀ 仅 33 µg mL⁻¹(图 3C)。铁/铜螯合剂(DFO、TTM、TEPA)可显著逆转杀伤,坏死或焦亡抑制剂无效,确认死亡依赖铁/铜。
为强化 Fenton 反应,研究人员利用 Warburg 效应:CoCl₂ 模拟缺氧后胞外乳酸仍高,证明 Hepa1-6 倾向糖酵解。CHCA 阻断 MCT 使乳酸滞留,pH 下降;Au 纳米酶耗糖产 H₂O₂,二者协同使 •OH 荧光最强(图 3D)。同时,Zn²⁺/Cu²⁺耗竭 GSH,CO 下调 Nrf2/HMOX1/NQO1,总抗氧化能力及 DPPH 清除率均降低。BODIPY-C11 和 MDA 显示脂质过氧化显著加剧(图 3E),提示铁死亡启动。
对于铜死亡,研究人员发现糖酵解被 CHCA 抑制后,细胞转向氧化磷酸化(OXPHOS),TCA 循环增强。2-DG 或半乳糖替代葡萄糖均使 Cu(II)-Elesclomol 毒性增加,且 TTM 可完全挽救(图 3F-J)。Seahorse 长程监测证实 CHCA 诱导的 OXPHOS 表型稳定,不易回弹(图 3K-N)。AuNPs 进一步耗糖,激活 AMPK 并上调铜转运蛋白 ATP7A/B、SLC31A1,低糖环境显著增强 Cu(II)-Elesclomol 抑制率(图 3O-P)。
线粒体膜电位检测显示 PEG@AuCZ@CC 组红荧光几乎消失;免疫荧光共定位(Pearson > 0.7)表明 DLAT 寡聚斑点集中于线粒体,金含量越高绿荧光越强,证实铜死亡强度增加(图 3Q-R)。Western blot 系统验证:铁死亡标志 TFR1↑、SLC40A1↓、GPX4↓、LC3-II↑、FTH1↓;铜死亡标志 FDX1↑、DLAT↑、LIAS↑、POLD1↑、ACO2↑(图 3T-U)。综上,研究人员证实 PEG@AuCZ@CC 通过同步重编乳酸与葡萄糖代谢,高效触发 Hepa1-6 细胞铁死亡与铜死亡协同杀伤(图3S)。
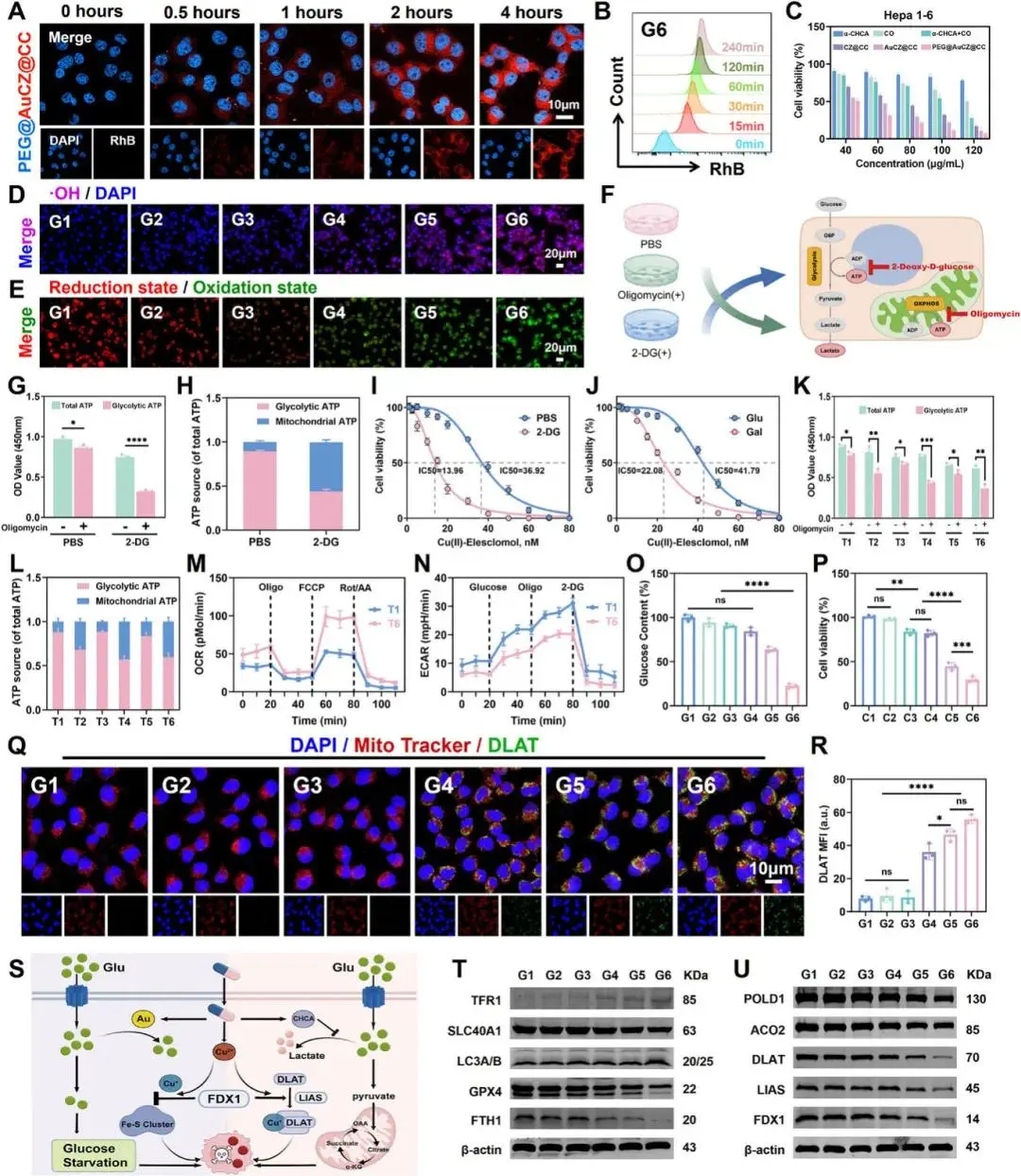
图3 PEG@AuCZ@CC NPs体外细胞摄取及抗肿瘤机制研究。A) Hepa1-6细胞与罗丹明B标记的PEG@AuCZ@CC NPs孵育不同时间后的CLSM图像,其中细胞核用DAPI染色。B) 流式细胞术分析罗丹明B标记的PEG@AuCZ@CC NPs的细胞摄取效率。C) 不同处理24小时后对Hepa1-6细胞的细胞毒性测定。D) 不同处理后羟基自由基染色的Hepa1-6细胞的荧光倒置显微镜图像,其中细胞核用DAPI染色。E) 不同处理后C11-BODIPY荧光探针染色的Hepa1-6细胞的荧光倒置显微镜图像。F) 寡霉素或 2-脱氧-D-葡萄糖处理 Hepa1-6 细胞后代谢物变化示意图。G) 不同处理后 Hepa1-6 细胞的 ATP 来源。H) Hepa1-6 细胞的主要代谢途径从糖酵解转向氧化磷酸化。I) 在用 Cu(II)-Elesclomol 处理的含有 PBS 或 2-DG 的培养基中生长的 Hepa1-6 细胞的活力。J) 在用 Cu(II)-Elesclomol 处理的含有高葡萄糖或半乳糖的培养基中生长的 Hepa1-6 细胞的活力。K) 不同处理后 Hepa1-6 细胞的 ATP 来源。L) 不同处理后 Hepa1-6 细胞的主要代谢途径从糖酵解转向氧化磷酸化。海马实验测量用 PEG@Au/Cu/ZIF-8@CHCA 重复处理的 Hepa1-6 细胞的 M) OCR 和 N) ECAR。O) 不同处理后 Hepa1-6 细胞中的葡萄糖含量。P) 在不同培养基中生长的 Hepa1-6 细胞的活力。Q) 代表性 CLSM 图像和 R) 不同处理后 Hepa1-6 细胞中 DLAT (绿色) 表达的半定量分析。细胞线粒体与 MitoTraker (红色) 共染色,细胞核与 Hoechst 33 342 (蓝色) 共染色。S) PEG@AuCZ@CC NPs 诱导的细胞内杯状凋亡示意图。T) 不同处理后 Hepa1-6 细胞中 TFR1、SLC40A1、LC3A/B、GPX4 和 FTH1 的蛋白质印迹分析。U)不同处理后 Hepa1-6 细胞中 POLD1、ACO2、DLAT、LIAS 和 FDX1 的蛋白质印迹分析
(4)PEG@AuCZ@CC体外抗肿瘤作用及其抑制增殖和迁移的潜在机制
研究人员首先通过 Calcein-AM/PI 双染证实 PEG@AuCZ@CC 的肿瘤杀伤效能:视野中几乎全部为红色死亡信号,活细胞绿色荧光极少(图 4A)。流式进一步定量,该组凋亡率显著高于其余各组(图 4B)。EdU 掺入实验显示,其红色增殖荧光最弱,表明 DNA 合成被显著抑制(图 4C);平板克隆结果亦显示集落形成能力最低(图 4D)。Transwell 迁移/侵袭及划痕实验共同表明,PEG@AuCZ@CC 可显著削弱 Hepa1-6 的运动能力(图 4E、F)。
为排除急性毒性干扰,研究人员将药物浓度降至亚致死剂量,发现细胞虽保持贴壁,却出现扁平、肿胀及核体积增大等形态改变,且 12–48 h 内密度无增加,提示增殖停滞。Western blot 显示 MMP2/9 下调,证实迁移力受抑(图 4G);流式检测显示细胞被阻滞于 G0/G1 期(图 4I、K)。机制上,G1/S 转换关键蛋白 p-RB、Cyclin E1、CDK4 表达降低,CDK2 升高,表明 checkpoint 被激活(图 4H)。彗星电泳可见明显 DNA 拖尾,而抗氧化剂 α-维生素 E 可显著减轻损伤并逆转 G0/G1 阻滞(图 4J–N),说明氧化应激介导的 DNA 断裂是周期停滞的主因。3D 肿瘤球模型进一步证实,PEG@AuCZ@CC 组球体数量与直径下降最为显著(图 4O)。综上,研究人员证明 PEG@AuCZ@CC 在低浓度即可通过氧化应激-DNA 损伤-G0/G1 阻滞途径持续抑制肝癌细胞增殖与转移,兼具直接细胞毒与细胞周期调控双重抗肿瘤效应,为克服实体瘤内药物浓度异质性提供了潜在解决方案。
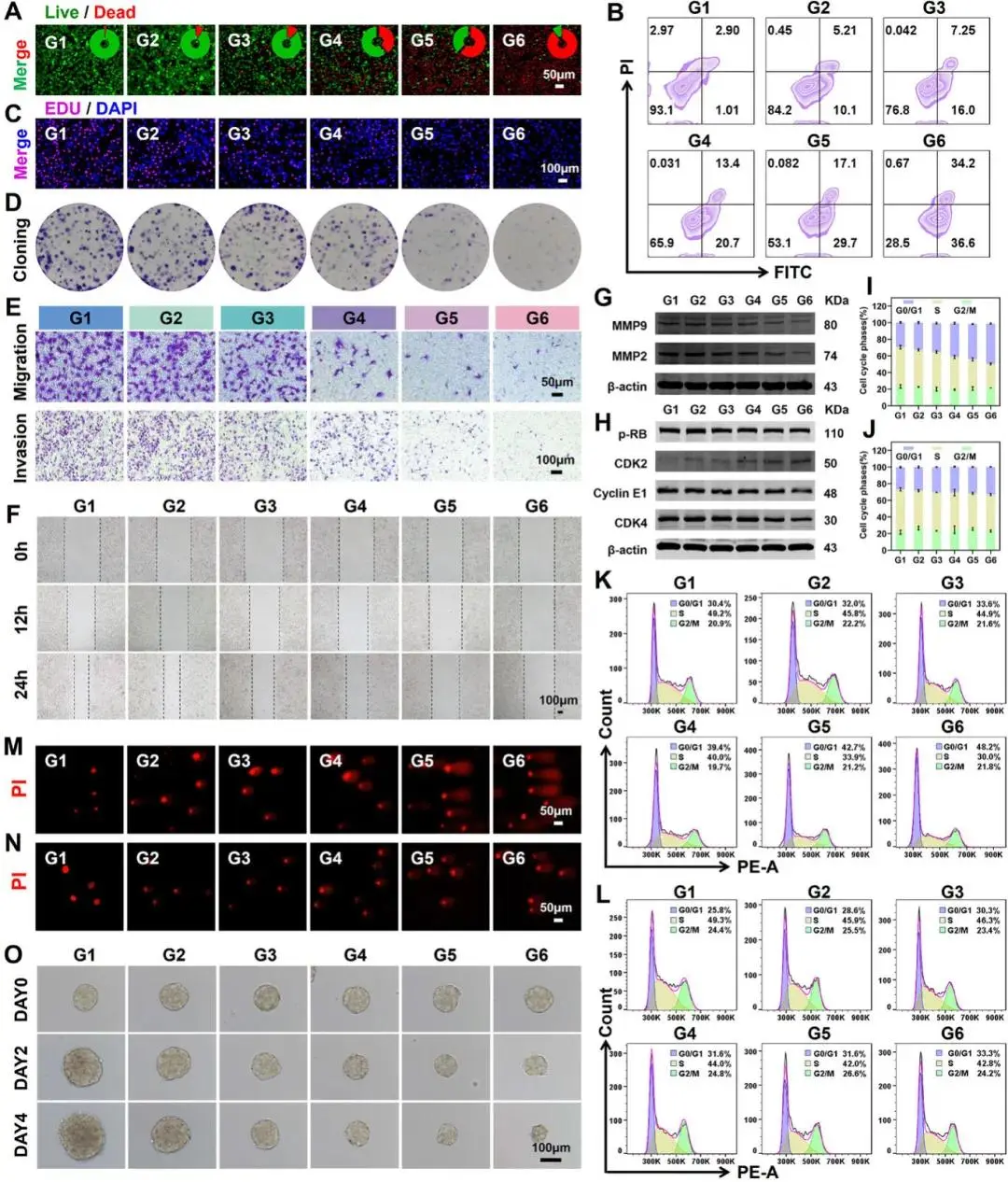
图4 PEG@AuCZ@CC体外抗肿瘤作用及其抑制增殖、迁移的机制研究。A) 碘化丙啶(PI)和钙黄绿素AM染色后Hepa1-6细胞的荧光倒置显微镜图像及半定量分析。B) 碘化丙啶(PI)和Annexin V-FITC检测试剂盒染色后Hepa1-6细胞的流式细胞术分析。C) EDU染色后Hepa1-6细胞的荧光倒置显微镜图像,其中细胞核用DAPI染色。D) 集落实验评估不同处理后Hepa1-6细胞的增殖情况。E) Transwell迁移和侵袭光学图像评估不同处理后Hepa1-6细胞的转移能力。 F) 处理后 0、12 和 24 小时的伤口愈合实验的明视野图像。G) 不同处理后 Hepa1-6 细胞中 MMP2 和 MMP9 的蛋白质印迹分析。H) 不同处理后 Hepa1-6 细胞中 p-RB、Cyclin E1、CDK2 和 CDK4 的蛋白质印迹分析。I,J) 不同处理后 Hepa1-6 细胞用碘化丙啶 (PI) 染色的定量数据和 (K, L) 流式细胞术模式。(M, N) 彗星试验评估不同处理后 Hepa1-6 细胞的 DNA 损伤。O) 不同处理后 Hepa1-6 细胞的肿瘤球体实验
(5)体外修饰免疫细胞功能
研究人员首先验证 PEG@AuCZ@CC 诱导的免疫原性细胞死亡(ICD)。免疫荧光显示,处理后 Hepa1-6 细胞膜 CRT 荧光最强,HMGB1 胞内残留弱,证实二者分别发生膜转位与分泌(图 5A-B;图 S44);Western blot 趋势一致(图 5C)。胞内 ATP 降至对照 36.33 %,培养基中升至 477.18 %,提示大量外排(图 5D-E)。随后获取 C57 小鼠骨髓原代 DC,与处理后的肿瘤碎片共培养 24 h:镜下见 DC 由圆形变为树突状成熟形态,流式示 CD80⁺CD86⁺ 成熟 DC 占 27.90 %,显著高于各对照(图 5G);ELISA 测得 IL-6、IL-12、TNF-α、IFN-γ 均显著升高,表明 ICD 有效启动 DC 成熟及促炎因子分泌。
在微环境层面,研究人员发现 PEG@AuCZ@CC 阻断 MCT1/2/4 后,培养基乳酸由 14.5 ± 0.3 mm 降至 9.2 ± 0.4 mm(图 5H)。建立 RAW264.7-Hepa1-6 共培养体系,流式与 Western 一致显示:该组 M2 标志 Arg-1、CD163/CD206 最低,M1 标志 iNOS、CD86 最高(图 5J-L;图 5K、M-O;);ELISA 示 IL-6、TNF-α 升高,抑瘤因子 IL-10 下降(图 5P-R),证实乳酸耗竭驱动 TAM 由 M2 向 M1 复极。
针对 Treg,研究人员先从 C57 脾分选 Treg,并在不同葡萄糖/乳酸浓度下培养 24 h。ELISA 显示乳酸变化对 IL-10/TGF-β 分泌影响远大于葡萄糖。将低乳酸条件培养的 Treg 与 CTLL-2 共培养,发现其对 CD4⁺/CD8⁺ IFN-γ⁺ 的抑制能力明显减弱。继而用 Transwell 将 Treg 与经药物预处理的 Hepa1-6 上清共培养:PEG@AuCZ@CC 组(G6)乳酸最低,IL-10/TGF-β 同步下降;CFSE 及流式显示 CTLL-2 增殖与 IFN-γ 分泌显著增强,表明乳酸减少直接削弱 Treg 抑制功能。综上,研究人员证实 PEG@AuCZ@CC 通过“耗乳酸-增 ICD-复极 M1-抑 Treg”级联,有效重塑免疫抑制微环境,为后续抗肿瘤免疫治疗提供坚实基础。
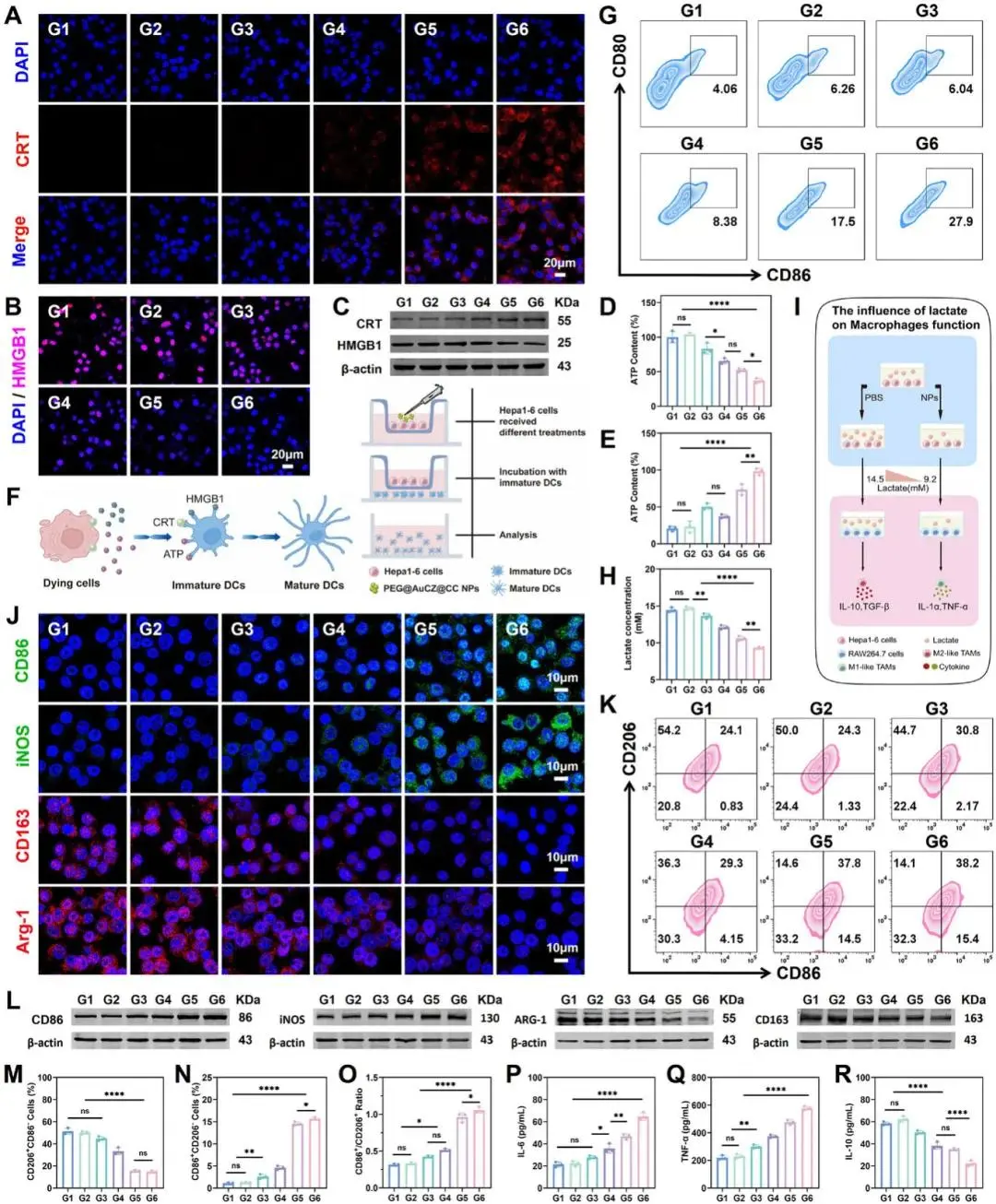
图5 体外修饰免疫细胞功能。A) 不同处理后Hepa1-6细胞膜上CRT定位的CLSM图像,其中细胞核用DAPI染色。B) 不同处理后Hepa1-6细胞核内HMGB1定位的CLSM图像,其中细胞核用DAPI染色。。C) 不同处理后Hepa1-6细胞中CRT和HMGB1的Western印迹分析。D) 不同处理下Hepa1-6细胞胞内ATP水平;E) 胞外ATP水平。F) 未成熟树突状细胞(DC)与Hepa1-6细胞共培养体系示意图。G) 不同处理组DC成熟情况的流式细胞术分析。(H) 不同处理后24小时Hepa1-6细胞培养基中乳酸水平的测定。 I) 体外实验设计,探讨乳酸对 RAW264.7 细胞功能的影响。J) 抗 CD86、抗 iNOS、抗 CD163 和抗 Arg-1 抗体处理后 Hepa1-6 细胞的 CLSM 图像。K) 流式细胞术分析;M-O) 不同处理组 RAW264.7 细胞的定量数据。L) 不同处理后 RAW264.7 细胞中 CD163、Arg-1、CD86 和 iNOS 的蛋白质印迹分析。不同处理组细胞因子的分泌水平,包括 P) IL-6、Q) TNF-α 和 R) IL-10
(6)体内分布、生物安全性和体内抗肿瘤作用
研究人员首先确认了PEG@AuCZ@CC 的静脉安全性:与红细胞 37oC共孵 4 h,溶血率可忽略(≤1.2 %),RBC 形态完整。随后以 Hepa1-6-LUC 皮下瘤模型评估体内行为。Cy5.5 标记示踪显示,尾静脉注射 0.5 h 后肿瘤荧光逐渐增强,PEG@AuCZ@CC 组 24 h 信号强度显著高于 AuCZ@CC 组(图 6A);离体分布证实肿瘤富集最高,肺、肝次之。延长观察至 48 h,肺信号 36 h 已明显衰减,48 h 消失,而肿瘤仍高亮,排除肺毛细血管滞留造成的假阳性。对肺上皮(MLE-12)与血管平滑肌(MOVAS)细胞毒性试验显示,120 µg mL⁻¹ 作用 48 h 存活率 >75 %,提示肺蓄积无明显毒性。治疗实验设 6 组(Control、CO、CO+CHCA、CZ@CC、AuCZ@CC、PEG@AuCZ@CC),第 1、4、7 天尾静脉给药,共 3 次(图 6C)。给药期间小鼠体重波动 <5 %(图 6I)。21 天处死可见,PEG@AuCZ@CC 组肿瘤体积最小,抑瘤率达 85 %(图 6D-H),且显著延长生存期。H&E 与 TUNEL 显示该组出现大面积核碎裂、溶解及阳性凋亡信号;Ki67 荧光最弱,提示增殖被显著抑制。机制层面,免疫荧光与 Western blot 同步证实:瘤内 PTGS2 上调、GPX4 下调,DLAT 聚集成斑,表明铁死亡与铜死亡共同执行杀伤(图 6J)。伴随 ICD 标志,CRT 膜曝光增强,HMGB1 由核转胞外(图 6K);引流淋巴结 DC 成熟率(CD80⁺CD86⁺)升至 28.7 %,显著高于对照。综上,研究人员阐明 PEG@AuCZ@CC 经静脉给药可安全富集于肿瘤,通过同步触发铁死亡/铜死亡-ICD-DC 成熟轴,实现高效体内抗肿瘤效应。
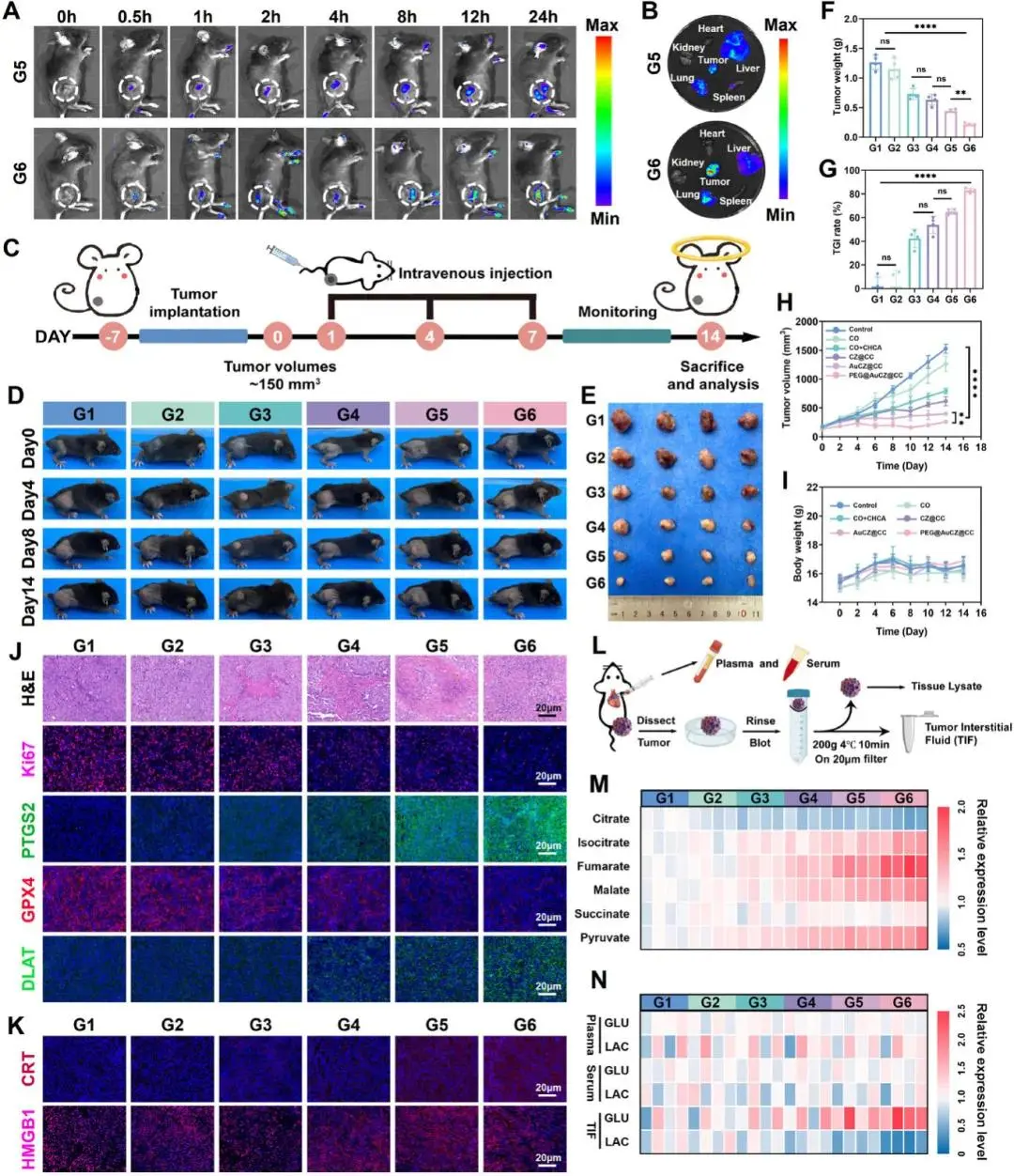
图6 PEG@AuCZ@CC NPs的体内抗肿瘤评价。A) 注射Cy5.5标记的AuCZ@CC NPs和Cy5.5标记的PEG@AuCZ@CC NPs后,Hepa1-6荷瘤小鼠体内随时间变化的荧光图像。B) 注射后24小时采集的肿瘤和正常器官的离体荧光图像。C) 体内抗肿瘤流程示意图。D) G1-G6组中第0天、第4天、第8天和第14天接受不同治疗的Hepa1-6荷瘤小鼠的照片。E) 不同治疗后肿瘤的数码照片。F) G1-G6组中第14天采集的肿瘤重量。(G) 各组肿瘤生长指数(TGI) 。H) G1-G6组中接受相应治疗的各组肿瘤体积随时间的变化。I) G1-G6组中小鼠体重随时间的变化。 (J) 第14天G1-G6期肿瘤切片的H&E、Ki67免疫荧光图像、PTGS2免疫荧光图像、GPX4免疫荧光图像和DLAT免疫荧光图像。K) 不同处理后肿瘤组织中CRT和HMGB1的免疫荧光图像。L) 研究肿瘤中的代谢重编程及其对肿瘤微环境的影响。M) 不同处理组间氧化磷酸化和糖酵解相关中间代谢物表达的变化。(N)不同处理后血清、血浆和肿瘤间质液中的葡萄糖和乳酸水平
(7)体内抗肿瘤免疫治疗
研究人员在体内验证 PEG@AuCZ@CC 的代谢重编程与免疫微环境重塑效应(图 6L)。肿瘤组织代谢组显示,该组氧化磷酸化中间产物上调、糖酵解中间产物下调,证实能量代谢向 OXPHOS 转移(图 6M)。同步采集血清、血浆及瘤内液(TIF)发现,TIF 中乳酸由 14.5 ± 0.3 mm 降至 9.2 ± 0.4 mm,葡萄糖升高;外周血变化不明显,说明重编程主要发生于瘤内(图 6N)。
代谢改善带来免疫抑制细胞锐减:流式示 Treg 占比由 41.0 % 降至 13.2 %,MDSC 由 47.0 % 降至 18.0 %(图 7A-B)。IHC 与流式共同表明 M2 标志 CD163/Arg-1 下调、M1 标志 iNOS/CD86 上调,M1/M2 比值显著升高(图 7C-D)。伴随 ICD 与 mDC 激活,瘤内 CD4⁺ 与 CD8⁺ T 细胞浸润增加,且 IFN-γ⁺ 比例升高(图 7E-G);脾脏效应记忆 T(CD8⁺CD44⁺CD62L⁻)提高 2.9 倍(图 7H)。瘤内促炎因子 IL-1α、IL-12p70、IL-6、TNF-α、IFN-γ 升高,抑炎 IL-10、TGF-β 下降(图 7I-O)。为排除炎症反应源于全身毒性,研究人员建立 LPS 急性(5/10 mg kg⁻¹)与亚急性/慢性模型(0.3 mg kg⁻¹ 连续注射 + 棉球肉芽肿)。PEG@AuCZ@CC 组小鼠体重、行为评分稳定,血清 IL-6、TNF-α、IL-1β 峰值远低于炎症对照;外周血 NLR、SII、SIRI 及 ALT/AST 与 PBS 组无差异,证实无系统炎症。长期安全评价延伸至 30 天:H&E 显示心、肝、脾、肺、肾无组织学损伤;血常规、肝肾功能、心肌酶谱均在正常范围;ICP-MS 未检出上述器官 Zn、Cu 沉积。氧化应激指标(4-HNE、8-OHdG、SOD、CAT、GSH-Px、MDA)亦无异常,表明高代谢器官未受氧化损伤。综上,研究人员证实 PEG@AuCZ@CC 通过瘤内“耗乳酸-促 OXPHOS”代谢重编程,协同诱导 ferroptosis/cuproptosis-ICD,高效逆转免疫抑制微环境,激活 T 细胞应答并建立记忆,且无全身毒性,具备良好的临床转化前景。
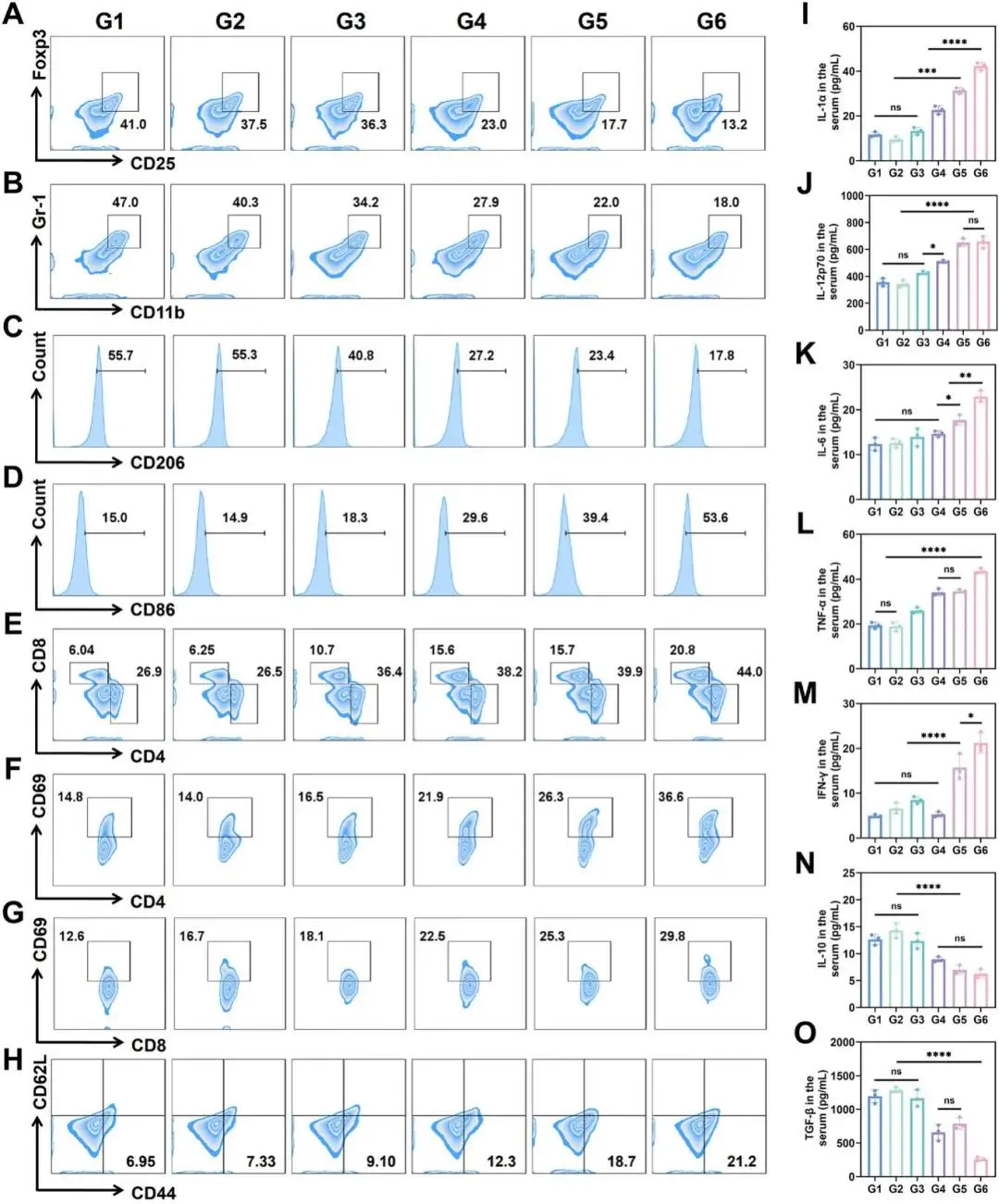
图7 体内抗肿瘤免疫治疗。A) 流式细胞术分析接受不同治疗的小鼠肿瘤中的Treg细胞。B) 流式细胞术分析肿瘤MDSCs。流式细胞术分析接受不同治疗的小鼠Hepa1-6肿瘤中的C) M2 TAMs和D) M1 TAMs。E) 流式细胞术分析接受不同治疗的Hepa1-6肿瘤中的CD4 + T细胞和CD8 + T细胞。流式细胞术分析接受不同治疗的Hepa1-6肿瘤中的F) CD4 + CD69 + T细胞和G) CD8 + CD69 + T细胞。H) 流式细胞术分析接受不同治疗的小鼠脾脏中的效应记忆T细胞。I–O) 不同治疗后小鼠血清中不同细胞因子分泌水平
总之,研究人员构建的PEG@AuCZ@CC纳米体系通过“代谢重编程-多重细胞死亡-免疫微环境重塑”三级联动,在肝细胞癌模型中实现85%抑瘤率,为ferroptosis/cuproptosis协同免疫治疗提供了可扩展的平台。然而,这一策略的深层机制与普适性仍需进一步拓展:首先,需解析纳米药物诱导的周期阻滞是否伴随衰老逃逸及其对长期免疫记忆的影响,以明确细胞命运决定与免疫监视之间的关联;其次,应系统阐明葡萄糖匮乏状态下AMPK-铜转运蛋白轴如何放大铜死亡信号,并验证该机制在缺乏Warburg效应的肿瘤中的适用性;此外,未来还将探索基于线粒体氧化磷酸化或氨基酸代谢的普适性干预策略,通过替代糖酵解依赖的调控路径,为包括肝癌在内的多种实体瘤提供更广泛、安全且高效的治疗选择。

|
创赛生物 提供高品质的医疗产品和服务 |
联系我们 |
产品中心 |
扫码关注
关注公众号 扫码加客服
|

