
IF:15.7《NC》靶向免疫治疗通过减少活化成纤维细胞和调节肺泡细胞分布来挽救肺纤维化
专栏:学术前沿
发布日期:2025-10-21
免特发性肺纤维化(IPF)是一种病因不明的纤维化间质性肺病,具有高发病率和死亡率。鉴于目前针对肺纤维化的治疗手段有限,众多研究致力于探索抗纤维化策略及其发病机制。成纤维细胞过度活化(以成纤维细胞活化蛋白FAP为标志)导致胶原纤维异常释放和过度堆积,引发细胞外基质(ECM)的病理改变及持续性炎症反应,形成细胞活动与细胞外纤维化相互作用的恶性循环。相较于传统阻断信号通路的药物,嵌合抗原受体(CAR)T疗法在纤维化治疗领域展现出广阔前景。近年来,脂质纳米颗粒(LNP)-mRNA系统被用于体内生成CAR-T细胞,有效抑制了癌症和心脏纤维化。然而,关于LNP-mRNA系统在肺纤维化治疗中的应用研究仍较为匮乏。

针对上述问题,上海交通大学霍云龙教授团队设计的LNP - mRNA系统在体内生成的瞬时FAP靶向CAR - T(FAPCAR - T)细胞的治疗活性,并探索在博莱霉素(BLM)诱导的肺纤维化小鼠模型中的肺再生机制。该团队开发了一种新型LNP-mRNA系统,并在博来霉素(BLM)诱导的肺纤维化小鼠模型中验证了其肺再生机制。研究假设认为,持续的纤维化积聚会通过支持炎症环境和改变细胞形态,导致细胞外基质(ECM)硬化并阻碍肺部再生。为验证该假说,该团队对BLM诱导肺纤维化的年轻/老年小鼠模型进行了蛋白质组学和单细胞测序(scRNA-seq)的全面分析。结果显示,ECM硬化会抑制II型肺泡上皮细胞(AT2)向I型肺泡上皮细胞(AT1)的分化,从而损害肺泡结构的自我修复能力。LNP-mRNA疗法不仅消除了肺纤维化,还通过改善ECM环境增强了AT2和Pclaf+细胞的可塑性,成功挽救了AT1细胞群。此外,肺部免疫功能通过Apoe+巨噬细胞重建,并伴随效应T细胞数量的增加而恢复。总体而言,优化后的LNP-mRNA系统在体内成功生成FAPCAR-T细胞,清除肺纤维化病灶,并促进肺部修。该文章于2025年4月21日以《Targeted immunotherapy rescues pulmonary fibrosis by reducing activated fibroblasts and regulating alveolar cell profile》为题发表于《Nature communications》(DOI:10.1038/s41467-025-59093-7)。
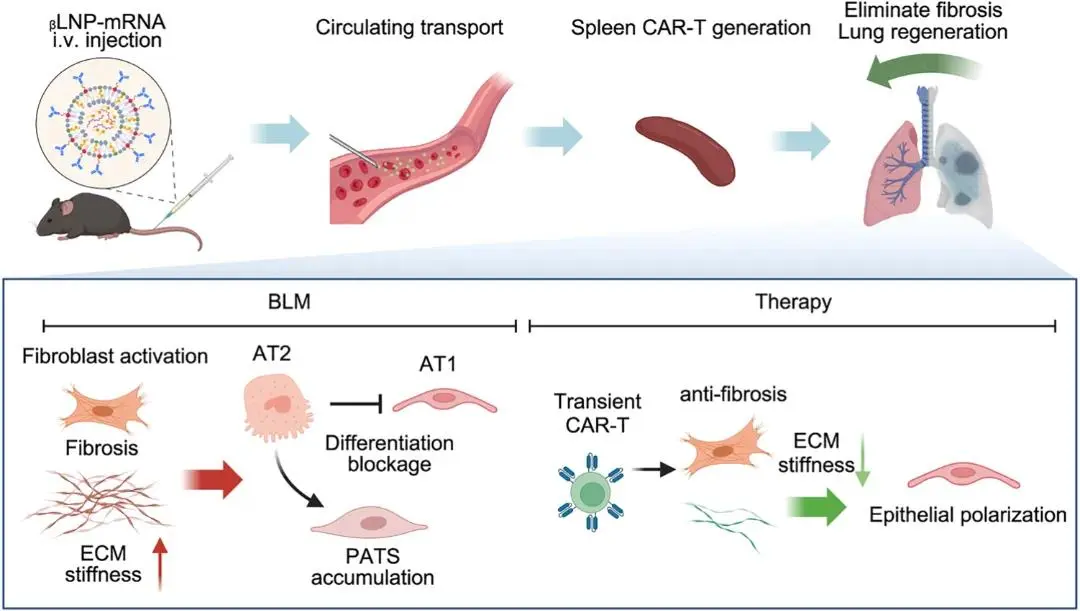
图1. LNP-mRNA治疗肺纤维化及相关机制
(1)通过CD5/βLNP-mRNA系统产生的FAPCAR-T细胞
通过用等量的β-谷甾醇替换胆固醇来提高转染效率(图2A)。先前的研究提出,β-谷甾醇可以促进脂质颗粒的内质网逃逸。不同比例的β-谷甾醇与LNP(0-40%)对LNP的物理特性影响不大。测试了在不同β-谷甾醇浓度下,经过24小时孵育后,293T细胞、成纤维细胞和正常肝细胞中荧光素酶mRNA的表达。含有30% β-谷甾醇和10%胆固醇的LNP表现出最高的mRNA表达,并被定义为βLNP。透射电子显微镜(TEM)扫描显示,βLNP和CD5修饰的βLNP(CD5/βLNP)之间没有显著差异,两者都具有约4纳米的双层膜和含有密集核苷酸的核心区域(图2B)。
体外转录(IVT)mRNA包含5'帽子结构、优化的UTR结构域、Kozak序列、修饰的poly(A)尾和编码CAR序列的ORF结构域(图2C)。除了传统的带有CD28跨膜结构域(TMD)的CAR设计外,还引入了一种新的TMD设计,遵循可编程膜蛋白(proMP)策略。先前的研究表明,来自CD28的跨膜片段有可能招募额外的共刺激信号,这可能导致CAR-T细胞的过度激活。这种效应在癌症治疗中可能是有益的,但在非肿瘤应用中可能导致不必要的细胞激活和细胞因子释放。成纤维细胞激活蛋白(FAP)是激活成纤维细胞的标志物。在博莱霉素给药后14天和28天,检测到肺组织中FAP的高表达(图2D)。基于这些发现,设计了两种FAP靶向CAR序列,分别带有CD28 TMD(FAPCAR 2.1)和优化的TMD(FAPCAR 2.2)。
经过24小时共孵育后,CD5/βLNP-mRNA与原代小鼠T细胞的FAPCAR 2.1/2.2在T细胞膜上的表达率约为80%(图2E)。细胞毒性实验表明,与仅表达FAP的靶细胞共培养时,瞬时FAPCAR-T细胞的溶解能力取决于效应细胞与靶细胞的比例(E:T)(图2F)。此外,与CD5/βLNP-FAPCAR 2.1相比,CD5/βLNP-FAPCAR 2.2导致细胞因子释放减少了约30%,而没有改变溶解能力(图2G),表明优化的TMD的优越性,使其更适合用于肺纤维化。此外,CD5/βLNP主要在脾脏和肝脏中累积,而IgG/βLNP仅在肝脏中累积,这表明CD5/βLNP具有靶向脾脏的能力(图2H)。为了进一步验证这一点,在注射后24小时对小鼠的脾脏进行了分析,发现CD5/βLNP主要与T细胞(CD3e阳性)共定位,很少与B细胞(CD19阳性)或树突状细胞(DCs,CD11c阳性)共定位。此外,部分CD5/βLNP与F4/80阳性巨噬细胞共定位,这表明外源性LNP被巨噬细胞吞噬(图2I,S2D)。随后,对小鼠进行了不同剂量的荧光素酶mRNA表达分析。在脾脏和肝脏中,注射量越大,表达量越高;然而,这些器官中的表达在96小时后被系统性清除(图2J,S2E)。然后,通过流式细胞术分析了FAPCAR mRNA在体内的转染情况。结果显示,在注射10μg的CD5/βLNP-FAPCAR后24小时,约3%的原代T细胞被编程为FAPCAR-T细胞,这比生理盐水注射组显著增加(图2K)。CD5/βLNP-FAPCAR 2.2治疗在体内成功生成了FAPCAR-T细胞。
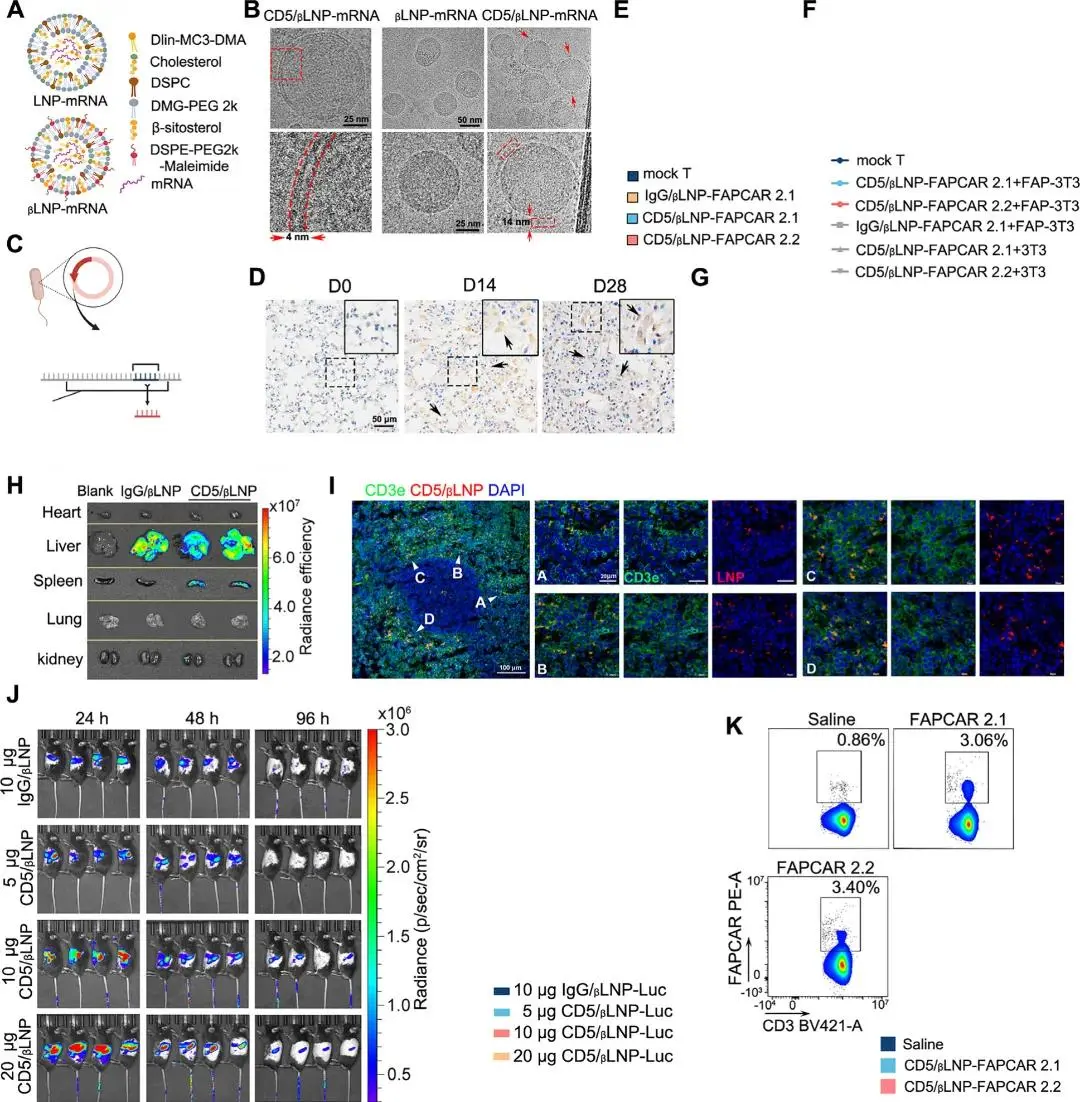
图2. A.传统和β-谷甾醇修饰的靶向LNP(βLNP)结构示意图。B. βLNP和带有CD5表面修饰的βLNP(STEM图像)的膜结构。C. FAPCAR 2.1和FAPCAR 2.2 IVT mRNA示意图。优化的TMD替换了FAPCAR 2.2序列中的CD28 TMD。D. 博莱霉素建模后0、14和28天小鼠肺中FAP的IHC染色。箭头:FAP阳性过度激活的成纤维细胞。E. 经过24小时与IgG/βLNP或CD5/βLNP共孵育后,小鼠T细胞表面FAPCAR 2.1和FAPCAR 2.2受体的表达。F. 经过36小时共培养后,瞬时CAR-T与3T3细胞的靶向溶解实验。G. 经过48小时共培养后,FAPCAR-T与FAP-3T3细胞的细胞因子产生(效应细胞:靶细胞比例=10:1)。 H. 注射后24小时,用PKH26标记的βLNP在小鼠脾脏和肝脏中的分布。I. 注射后24小时,脾脏中CD5/βLNP(红色)与CD3e阳性T细胞(绿色)的共定位。J. 不同剂量下,βLNP在小鼠体内转染后荧光素酶mRNA的剂量和时间依赖性分布和表达变化。K. 与生理盐水注射相比,注射10微克CD5/βLNP后24小时,通过流式细胞术检测FAPCAR 2.1和2.2 mRNA的表达
(2)CD5/βLNP-FAPCAR 2.2治疗逆转肺纤维化
通过CD5/βLNP-FAPCAR 2.2治疗成功在体内生成了FAPCAR-T细胞。随后在博莱霉素诱导的肺纤维化小鼠模型中评估了其抗纤维化反应。博莱霉素模型的小鼠分别接受CD5/βLNP-FAPCAR 2.2、IgG/βLNP-FAPCAR 2.2或生理盐水处理。治疗组还在术后第7天和第14天接受了商业基准药物吡非尼酮(PFD,100 mg/kg/天)的口服给药(图3A)。苏木精-伊红染色显示,从术后第7天起肺纤维化和肺泡异常开始出现,并随着纤维化的进展逐渐加重(图S3A)。CD5/βLNP-FAPCAR 2.2治疗后,小鼠体重和肺系数迅速恢复,存活率在所有接受博莱霉素手术的组中最高(图3B–D)。在CD5/βLNP组(而不是在生理盐水或PFD组)中,在术后第15天观察到CD3+T细胞的积累(图S3B)。为了观察转染情况,在FAPCAR mRNA中生成了tdTomato报告基因。在脾脏和纤维化的肺部,可检测到共表达CD3和tdTomato的T细胞(图3E),这为CD5/βLNP转染和FAPCAR-T细胞浸润纤维化肺组织提供了证据。此外,流式细胞术显示,CD5/βLNP-FAPCAR 2.2治疗后,小鼠脾脏和肺部的FAPCAR阳性T细胞数量增加(图3F,S3C,D),而IgG/βLNP治疗或健康小鼠中则没有。重要的是,CD5/βLNP组的FAP阳性成纤维细胞数量显著减少,而PFD治疗对这一指标没有影响(图3G,S3E)。根据肺纤维化评估的标准指南,术后第28天对动物进行微计算机断层扫描(CT)分析。影像学结果表明,博莱霉素+生理盐水组在肺部区域出现区域实变和弥漫性阴影,肺部的Hounsfield单位(HU)密度增加,而这些症状在CD5/βLNP-FAPCAR 2.2治疗后显著缓解(图3H)。重建的3D肺模型显示,治疗后纤维化组织(红色区域)显著减少,肺部空气量增加,表明功能恢复(图3I,S3F)。然后对肺部进行充分灌注后收集。整个肺部外观图像显示,纤维化肺部因组织损伤导致间质红细胞渗漏,出现红色病变,而健康组和CD5/βLNP治疗组的肺部呈白色(图3J)。
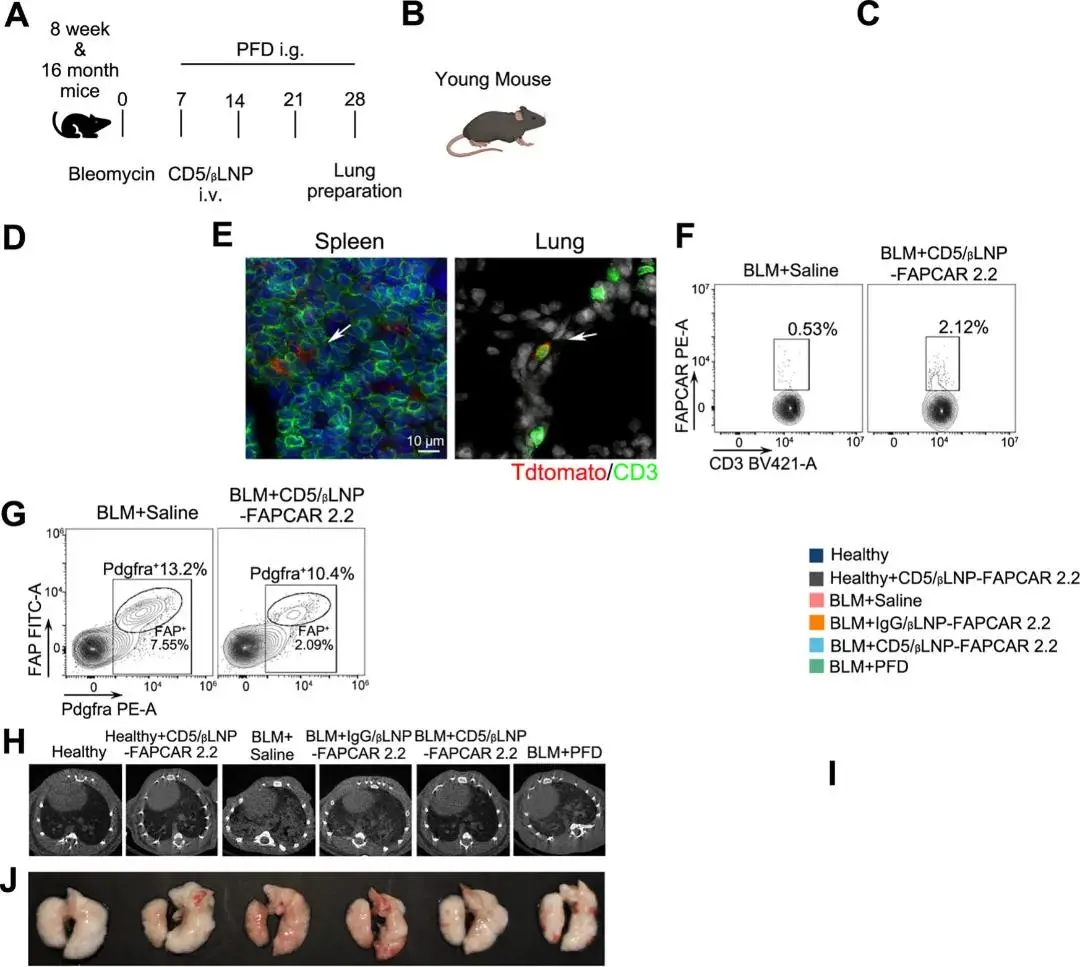
图3. A.年轻或老年小鼠纤维化模型建立及治疗策略示意图。B,C.年轻小鼠体重变化(B)及存活曲线(C)。D. 肺部指数。E.第二次注射CD5/βLNP-FAPCAR 2.2后24小时脾肺T细胞中tdTomato报告基因表达情况。F. 流式细胞术检测小鼠CD3+肺T细胞的表达比值。G. 流式细胞术分析小鼠肺部FAP+Pdgfra+成纤维细胞比例。H. 术后第28天的数字照片。I. CT图像和根据CT结果计算的肺部空气量和 J. 肺组织照片
(3)老年小鼠的抗纤维化反应
通过Masson和Picrosirius红染色评估显示,与生理盐水、IgG/βLNP或PFD组相比,CD5/βLNP治疗组的细胞外基质纤维化显著改善(图4A–C)。此外,羟脯氨酸(HYP)测定显示,经CD5/βLNP-FAPCAR 2.2治疗后,胶原蛋白含量接近健康水平(图4D)。机械测试评估显示,经CD5/βLNP-FAPCAR 2.2治疗后,全肺组织的弹性模量显著降低(图4E)。总之,注射CD5/βLNP-FAPCAR 2.2显著减少了FAP阳性细胞数量,并拯救了肺纤维化,而PFD治疗对纤维化的缓解效果有限,这可能是因为治疗时间较短。
特发性肺纤维化(IPF)的发病率与年龄呈正相关,且在老年人群中更为普遍。为了进一步验证CD5/βLNP-FAPCAR 2.2在抑制肺纤维化中的作用,在16月龄的老年小鼠中建立了纤维化模型。与年轻小鼠相比,老年小鼠在接受相同剂量的博莱霉素后,博莱霉素诱导的肺纤维化更为严重,表现为更大的区域实变面积、更高的Hounsfield单位(HU)密度以及更少的肺部空气量(图4I-K)。在接受CD5/βLNP-FAPCAR 2.2治疗后,与博莱霉素+生理盐水和IgG/βLNP组相比,老年小鼠的生存率显著提高,肺系数降低,尽管老年小鼠的体重恢复速度不如年轻小鼠快(图4F-H)。Masson染色和羟脯氨酸(HYP)定量显示,在CD5/βLNP-FAPCAR 2.2治疗后,老年小鼠的纤维化和胶原蛋白含量显著降低(图4L,M)。这些结果表明,CD5/βLNP-FAPCAR 2.2疗法可以抑制老年小鼠的肺纤维化。
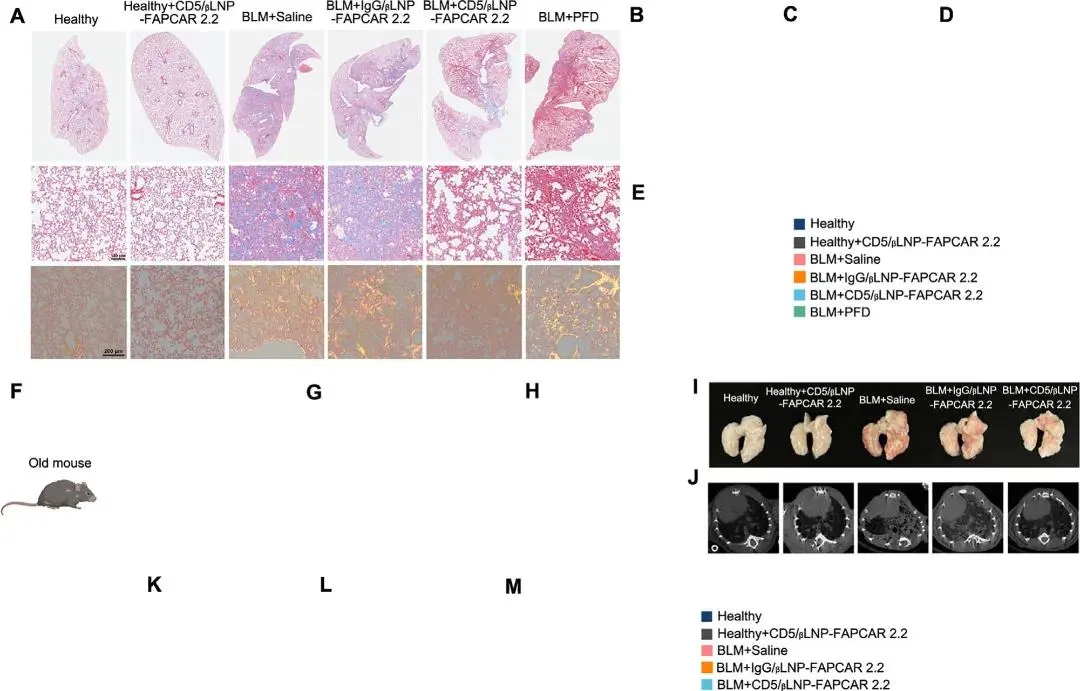
图4. A. Masson和Picrosirius红染色显示纤维化区域。B. 纤维化区域定量分析。 C. Ashcroft评分定量分析。D. 羟脯氨酸(HYP)测定用于定量胶原蛋白含量。E. 使用机械测试系统评估全肺组织的弹性模量。F. 老年小鼠体重变化。G. 老年小鼠存活率。H. 老年小鼠肺系数。I. 小鼠肺部照片。J,K. 微计算机断层扫描(CT)图像 。L. Masson染色定量分析纤维化区域。M. 羟脯氨酸(HYP)测定用于定量胶原蛋白含量
(4)纤维化肺中蛋白质的特征
对博莱霉素给药后0、14和28天的三个时间点进行了蛋白质组学分析(每组4只小鼠),结果显示BLM小鼠随时间推移蛋白质组发生了显著变化(图5A)。K-means聚类算法将所有组的4126个差异表达蛋白(DEPs)分为6个簇,这与热图结果一致。基于GOBP分析,簇被注释为DNA修复、免疫系统过程、细胞外基质(ECM)组织、细胞内信号转导、蛋白质运输和脂质代谢过程(图5B)。特别是,第3簇以ECM蛋白表达的持续增加为特征,这涉及到胶原蛋白和纤维连接蛋白表达的上调(例如,Fn1、Fbln1、Col4a2、Col18a1)在BLM给药后(图5C,D)。对因CD5/βLNP-FAPCAR 2.2治疗而恢复的肺与纤维化肺之间的蛋白质组学分析表明(每组4只小鼠),PCA揭示了BLM+生理盐水组和CD5/βLNP组之间DEPs的显著差异(图5E)。由CD5/βLNP-FAPCAR 2.2治疗下调的顶级DEPs富集在ECM、炎症和免疫调节中(图5F),例如下调的ECM相关蛋白(Lox、Ctsd和Ctsz)和上调的抗炎蛋白(Prx、Hpdg和Ifitm3)。这些结果表明,通过使用CD5/βLNP-FAPCAR 2.2,促进了抑制炎症环境和ECM重塑过程。
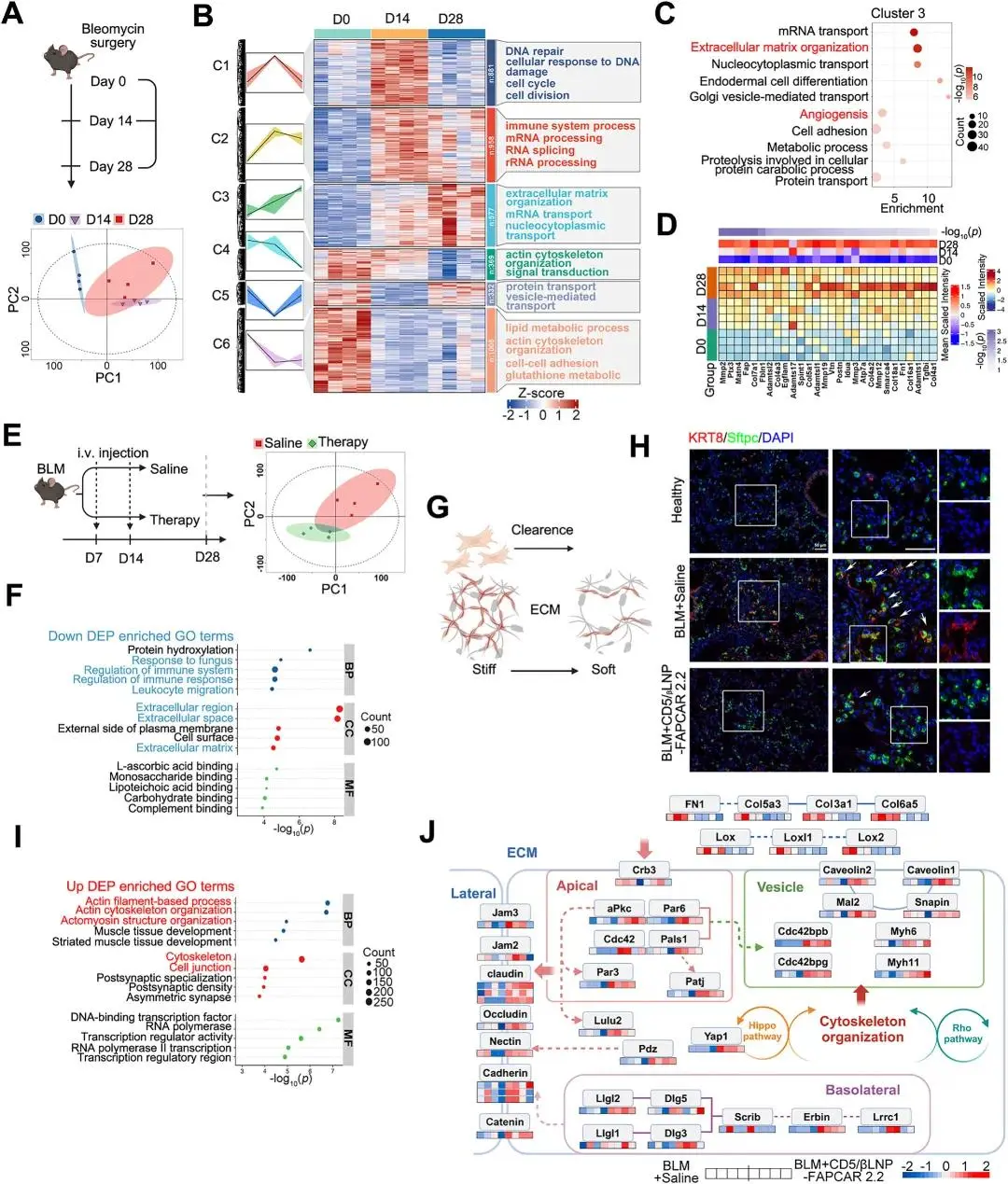
图5. A. 对纤维化组织进行蛋白质组学策略和PCA分析。B. 通过K-means算法获得的蛋白质簇,并在热图中显示,其中4126个差异表达蛋白被富集到6个簇中。基于GOBP分析对簇进行注释。C. 点图显示了第3簇中富集的顶级10个GOBP术语。D. 显示了博莱霉素给药后0、14和28天的ECM相关蛋白。E. 对治疗的BLM小鼠进行蛋白质组学策略和PCA分析。F. 点图显示了与图(E)对应的下调DEPs富集的顶级5个GOBP、GOCC和GOMF术语。G. 治疗前后纤维化清除和ECM僵硬度调节的示意图。H. 代表性免疫荧光图像(红色:KRT8;绿色:Sftpc;蓝色:DAPI)。I. 点图显示了与图(E)对应的上调DEPs富集的GO术语。J. 上皮极化途径的网络投影图。K. 代表性免疫荧光图像显示极化因子Cdc42和Pkcz(红色:Cdc42;绿色:Pkcz;蓝色:DAPI)在细胞中的位置。L. 代表性免疫荧光图像显示调节上皮极化的关键分子(蓝色:DAPI;红色:Calponin;绿色:Pkcz;白色:Cldn5)。M. 与血管生成(Vegfa、Eng)和二氧化碳调节(Ca4、Ca13)相关的蛋白质相对丰度
(5)抗纤维化治疗后肺泡上皮极化
蛋白质组学分析显示,在CD5/βLNP-FAPCAR 2.2治疗后,细胞外基质(ECM)中的纤维化被清除。机械测试评估显示,治疗后肺组织的弹性模量显著降低(图4E)。这些发现表明,细胞外组织张力降低(图5G),有助于AT2向AT1的分化。此外,免疫荧光结果显示,在治疗后PATS的数量显著减少(图5H),表明AT2向AT1的分化已恢复正常。AT2向AT1细胞的分化与细胞形状和结构的显著变化相关,通常被认为是上皮极化过程。这种转变通常伴随着信号和结构蛋白表达的许多变化。在CD5/βLNP-FAPCAR 2.2治疗后,上调的DEPs主要富集在细胞骨架和细胞间连接信号通路中,所有这些都与细胞极化相关(图5I),因此,根据KEGG分析绘制了极化分子图(图5J)。上皮细胞被认为是肺恢复途径富集的关键贡献者。
为了验证这一假设,需要排除巨噬细胞极化或上皮-间充质转化(EMT)过程,因为这两种过程可能在疾病进展过程中发生。通过蛋白印迹分析了EMT标记物的表达(图6A)。从上皮细胞转化而来的间充质细胞表达低水平的E-钙粘蛋白(一种粘附因子)和高水平的波形蛋白。这些标记物的表达在CD5/βLNP-FAPCAR 2.2治疗后接近正常水平。因此,治疗抑制了EMT,并且没有对极化信号富集做出贡献。另一方面,在蛋白质组学数据中,与巨噬细胞相关的富集基因在治疗后减少(图6B)。与基因表达的减少一致,流式细胞术结果显示,在炎症性纤维化肺中,标记为F4/80的巨噬细胞数量增加,而在CD5/βLNP-FAPCAR 2.2治疗后减少(图6C)。亚型分析显示,治疗减少了被鉴定为促纤维化表型的M2巨噬细胞的数量(图6D,E)。总体而言,巨噬细胞总数和M2亚型的减少排除了CD5/βLNP-FAPCAR 2.2治疗后巨噬细胞极化对途径富集的贡献。
对于上皮细胞极化,由Cdc42和aPKC(Pkc-zeta)形成的信号轴被认为是调节极化的关键。ECM信号通过Crb3感知并传递,Crb3招募Cdc42-aPKC-PAR6在顶膜区域形成复合体。这个复合体是将蛋白质从顶膜区域排除的关键步骤,通过调节细胞骨架,促进蛋白质在顶膜、侧膜和基底膜之间的极化分布。为了验证这一机制,通过蛋白印迹检测了Cdc42的表达水平。在CD5/βLNP-FAPCAR 2.2治疗后,肺组织中Cdc42的表达增加(图6A)。此外,免疫荧光结果显示,Cdc42和PKCz在细胞膜上共定位,并且这种共定位倾向于在细胞膜的一侧而不是均匀分布在细胞膜上(图6F,G)。细胞骨架相关的紧密连接得到增强(图6H)。此外,血管生成标记物(Eng,Vegfa)和二氧化碳转运蛋白(Ca4,Ca13)的上调(图6I)表明,蛋白质极化的增加驱动了受损结构和功能的BLM肺在清除过度激活的成纤维细胞后通过FAPCAR 2.2 T细胞的自我修复。
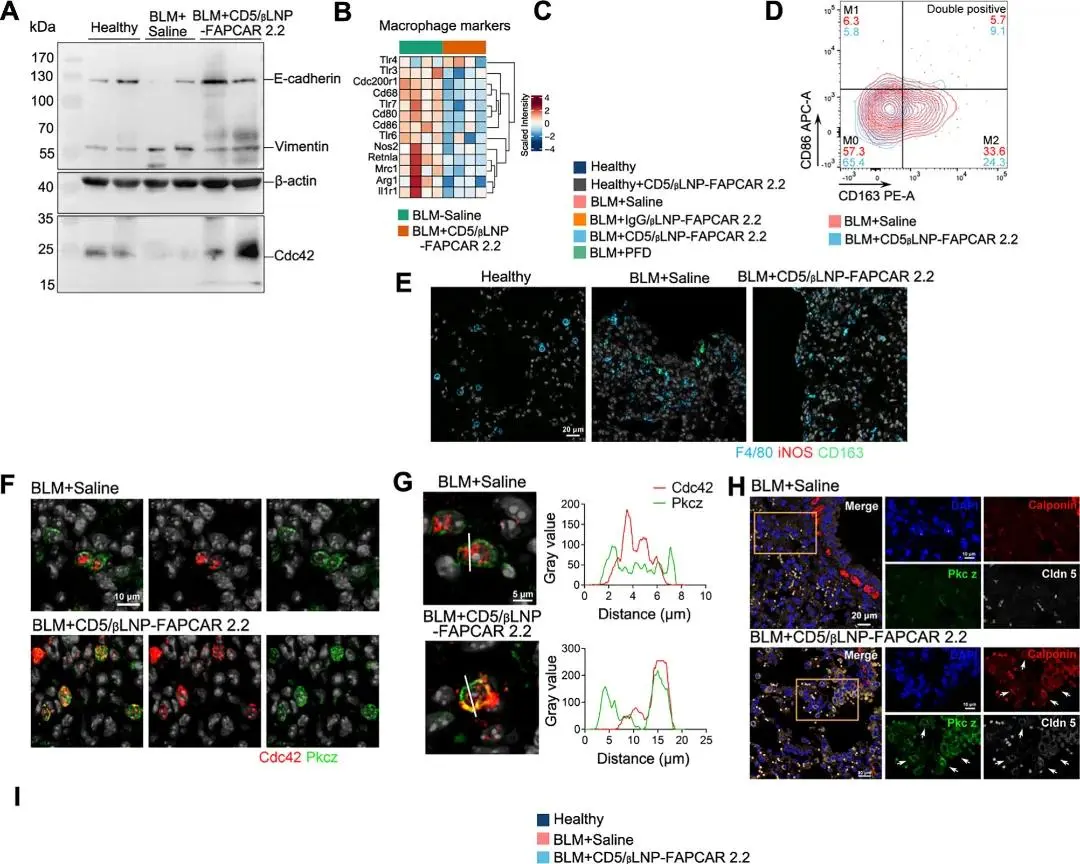
图6. A. 蛋白印迹图像和EMT标记基因以及Cdc42蛋白表达的定量。B. 巨噬细胞标记物的蛋白质组学热图。C. 流式细胞术分析显示肺细胞中巨噬细胞的百分比。D. 流式细胞术图显示M1/M2表型在BLM+生理盐水和CD5/βLNP组之间的比较。E. 代表性免疫荧光图像显示肺组织中的巨噬细胞。(青色:F4/80;红色:iNOS;绿色:CD163;白色:DAPI)。F,G. 代表性免疫荧光图像显示极化因子Cdc42和Pkcz(红色:Cdc42;绿色:Pkcz;白色:DAPI)在细胞中的位置。H. 代表性免疫荧光图像显示调节上皮极化的关键分子。(蓝色:DAPI;红色:Calponin;绿色:Pkcz;白色:Cldn5)。I. 与血管生成(Vegfa,Eng)和二氧化碳调节(Ca4,Ca13)相关的蛋白质相对丰度
(6)肺再生的细胞特征
蛋白质组学结果表明,蛋白通路富集在细胞外基质重塑和上皮极化中,暗示多种细胞类型可能参与信号调节。为了探索成功清除纤维化后的肺再生过程,通过10×Genomics平台对BLM+生理盐水和CD5/βLNP组的肺进行了单细胞测序分析(图7A)。通过建立的细胞标记物对捕获的细胞进行无偏分类和鉴定,将47,129个细胞分为21个簇。发现大多数细胞属于免疫系统,包括T细胞(CD3e+)、NK细胞(Nkg7+)、B细胞(CD79a+)和免疫单核细胞(Cst3+)。此外,还鉴定出几个小群体为肺实质细胞,包括肺泡细胞(Sftp+)、内皮细胞(Pecam+)和成纤维细胞(Dcn+)(图7B)。与BLM组相比,CD5/βLNP组中肺泡细胞、成纤维细胞、T细胞和巨噬细胞的数量发生了显著变化,表明CD5/βLNP-FAPCAR 2.2治疗调节了细胞特征(图7C,D)。
成纤维细胞分为三个不同的簇,包括炎症性、纤维化性和间皮细胞,这与最近的研究一致。簇0被鉴定为炎症性成纤维细胞,其高表达Cxcl12和Cxcl13。纤维化性成纤维细胞以Fbln2和其他病理性的细胞外基质基因特征。值得注意的是,两个过表达Col1a1和Col3a1的簇在治疗后显著减少。此外,间皮性成纤维细胞以Msln为特征,在所有组中均保持不变(图7E,F)。此外,FAP在纤维化性和炎症性簇中均有表达。这两个簇中的大多数细胞在BLM组中被发现,表明FAPCAR-T细胞有效地清除了目标细胞,而没有影响其他细胞群。
肺泡细胞是肺再生的关键因素(图7H)。AT1细胞被分为两个簇,即“AT1”,其Ager表达水平较高,以及“Hopxhigh AT1”,其Hopx表达特异性较高。Hopx参与早期发育和分化途径。就这个簇中的Krt8表达而言,Hopxhigh AT1被认为是一种中间状态。此外,簇2被鉴定为PATS,其高表达Krt8和Sftpc。Cxcl2+细胞群的特征是高表达与炎症、纤维化和脂质代谢相关的基因。这个簇在治疗后几乎消失,表明这些细胞可能是高度炎症性损伤细胞,这些细胞在肺纤维化发展过程中暂时出现(图7I-K)。上述所有被鉴定为中间状态或与炎症相关的簇在CD5/βLNP-FAPCAR 2.2治疗后减少,与蛋白质组学分析一致(图7I)。重要的是,在整个肺细胞的UMAP中鉴定出簇12为增殖性肺泡祖细胞(PAPCs)(图7B),这是一个与肺泡细胞相关的显著增加的簇。这一群体以Pclaf、Mki67和Top2a的特异性表达为特征(图7L)。先前的研究强调,PAPCs在肺泡细胞系可塑性中发挥关键作用,作为祖细胞库,并且对于肺再生是必不可少的。为了验证PAPCs的功能,重新聚类PAPCs与肺泡细胞(图7M),并使用RNA速度和Slingshot进行细胞系轨迹推断。PAPCs的发育轨迹被发现朝向肺泡细胞(图7N)。此外,AT1细胞是AT2、Hopxhigh AT1和PATS簇分化的终点。这些结果表明,在肺恢复过程中,肺泡细胞系可塑性增加,AT2或PAPCs在分化为AT1细胞中发挥了积极作用。
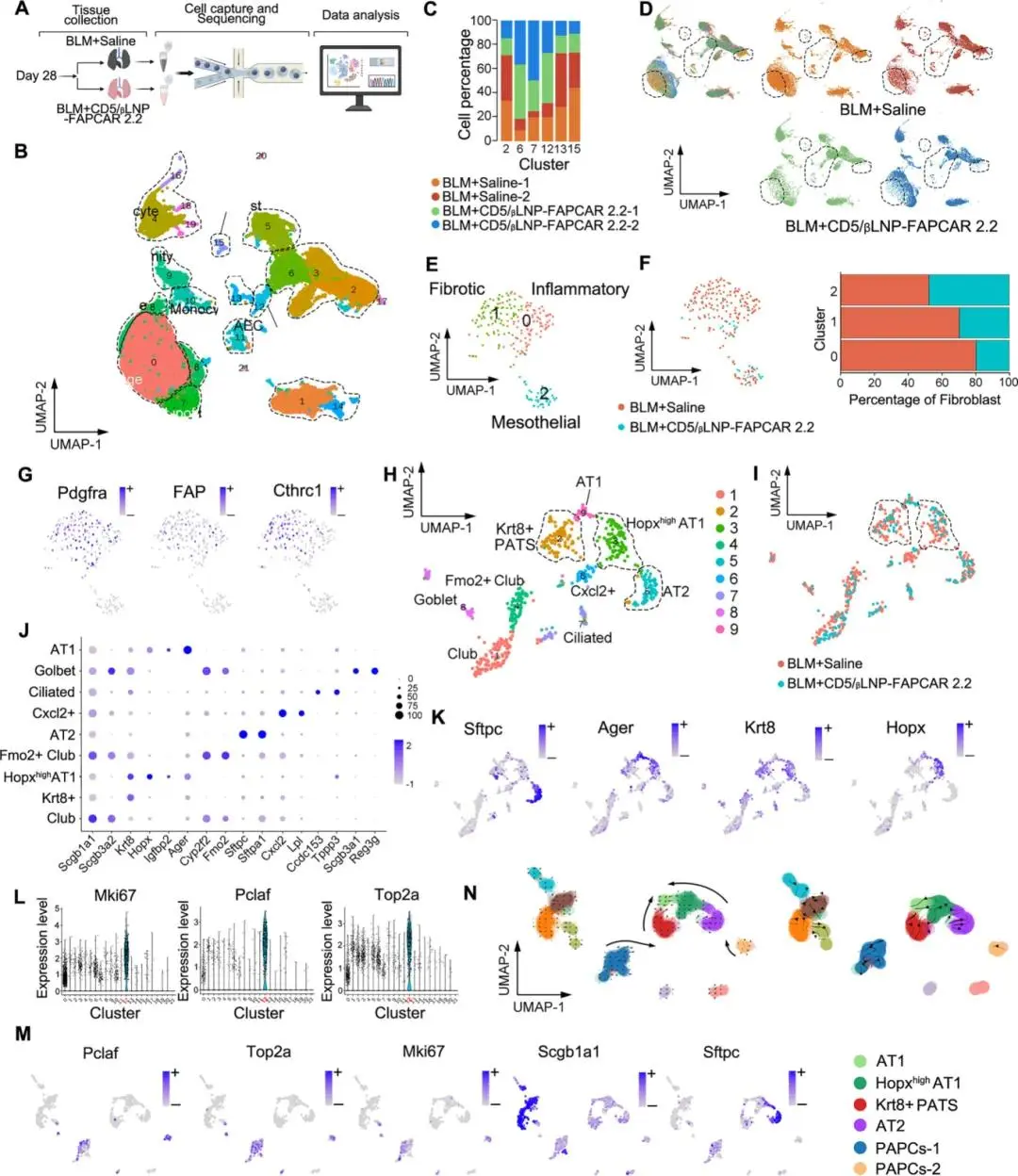
图7.A.小鼠肺单细胞测序研究设计。B. 细胞簇的UMAP图和对部分捕获的肺细胞的注释。C. 比较BLM+生理盐水和BLM+CD5/βLNP-FAPCAR 2.2组的簇成分。D. 显示显著变化的簇的样本分布。E. 成纤维细胞簇的UMAP嵌入。F. 比较BLM+生理盐水和BLM+CD5/βLNP-FAPCAR 2.2组的成纤维细胞细胞簇。G. 在UMAP上表达纤维化和过度激活的成纤维细胞标记基因的表达投影。H,I. 肺泡细胞的UMAP图和注释(H)以及比较BLM+生理盐水和BLM+CD5/βLNP-FAPCAR 2.2组的簇状态(I)。J. 每个肺泡细胞簇的代表性标记物的点图。K. 在整个肺细胞簇上表达特定肺泡上皮细胞标记物的表达投影。L, M. PAPCs标记基因在整个肺细胞簇中的表达(L)以及在与肺泡细胞重新聚类的UMAP图中突出显示PAPCs标记基因(M)。PAPCs:增殖性肺泡祖细胞。N. RNA速度分析包括肺泡细胞和PAPCs的簇
(7)Apoe+巨噬细胞和T细胞类型对肺免疫重建至关重要
除了结构恢复外,免疫重建也是器官再生的重要步骤。分析了巨噬细胞和T细胞。首先,巨噬细胞被分为10个簇,其中4个簇(簇2、4、5、7)在CD5/βLNP-FAPCAR 2.2治疗后显著减少,而3个簇(簇0、3、8)增加。簇0、1、2、4、5和6富含肺泡巨噬细胞(AM)标记物(Ear1、Ear2、Chil3、Plet1)(图8A-C)。其余簇为单核细胞来源的巨噬细胞(mono-M),表达Ccr5、CD33、Sell和Itgam。簇2、4和5被鉴定为促纤维化的脂质相关巨噬细胞(LAMs),因为它们高表达促纤维化基因(Fn1、Spp1、CD44)和脂质代谢基因(Lpl、Trem2、Fabp4、Fabp5)。簇7和9表达炎症基因(Il1b、S100a9、S100a8)和典型的骨髓标记基因(CD33),表明它们的单核细胞谱系(图8C)。簇7也参与促纤维化过程。减少的巨噬细胞(簇2、4、5、7)表明,FAPCAR-T细胞通过清除过度激活的成纤维细胞,打破了炎症-纤维化循环,这与流式细胞术和蛋白质组学结果一致。
由于ApoE是一种分泌蛋白,评估了细胞间通讯。CellphoneDB分析显示肺泡巨噬细胞与ApoE+巨噬细胞之间存在强烈的相互作用(图8D)。Trem2已被报道为触发髓系细胞分化和发育的受体。蛋白质相互作用分析显示,ApoE和Trem2配体对仅出现在簇1/3组中(图8E)。这些发现表明,ApoE+巨噬细胞可能有助于刺激髓系细胞的分化和发育。为了进一步确认这一发现,伪时间分析显示,表达高单核细胞标记物的簇作为根细胞,巨噬细胞通过中间簇3分化为产生成熟的先天免疫细胞或成熟的肺泡巨噬细胞(图8F)。相应地,RNA速度分析显示了从ApoE+单核细胞到AM簇的发展路径(图8G)。总体而言,与纤维化相关的肺泡巨噬细胞在CD5/βLNP-FAPCAR 2.2治疗后显著减少,而ApoE+单核细胞群体参与了肺泡巨噬细胞数量的定量补充,以维持其表型用于免疫重建。
对于T细胞,簇7-9被鉴定为成熟的效应细胞群体(Tc1和Th1)和CD4+记忆T细胞(Tm),通过标记基因进行注释。重新聚类显示,在CD5/βLNP-FAPCAR 2.2治疗后,这3个簇的细胞数量显著增加。幼稚T细胞(Tn,簇0、1、6)被鉴定为Lef1、Ccr7和Sell的特征,并且在BLM组中更为丰富(图8H-J)。KEGG富集显示,Tn细胞富含Jak-Stat通路和干细胞分化,而效应细胞群体富含Rap1信号通路、细胞因子释放通路和TCR激活通路(图8K)。基于伪时间、Slingshot和RNA速度,Tn细胞可以被分类为干细胞。这些分析进一步揭示了Tn细胞的不同发育轨迹:分化为高表达Ifng和Gzma的群体,或分化为Th17和γδ T细胞(图8L-N)。此外,Ly6a(TCR依赖性激活的标记)在Th1细胞中高表达。这些结果表明,由于CD5/βLNP-FAPCAR 2.2治疗在纤维化肺中招募了瞬时FAPCAR-T细胞,导致大量效应细胞在肺组织中积累。值得注意的是,这种治疗导致了记忆T细胞新簇的出现,而在BLM组中完全缺失(图8I)。
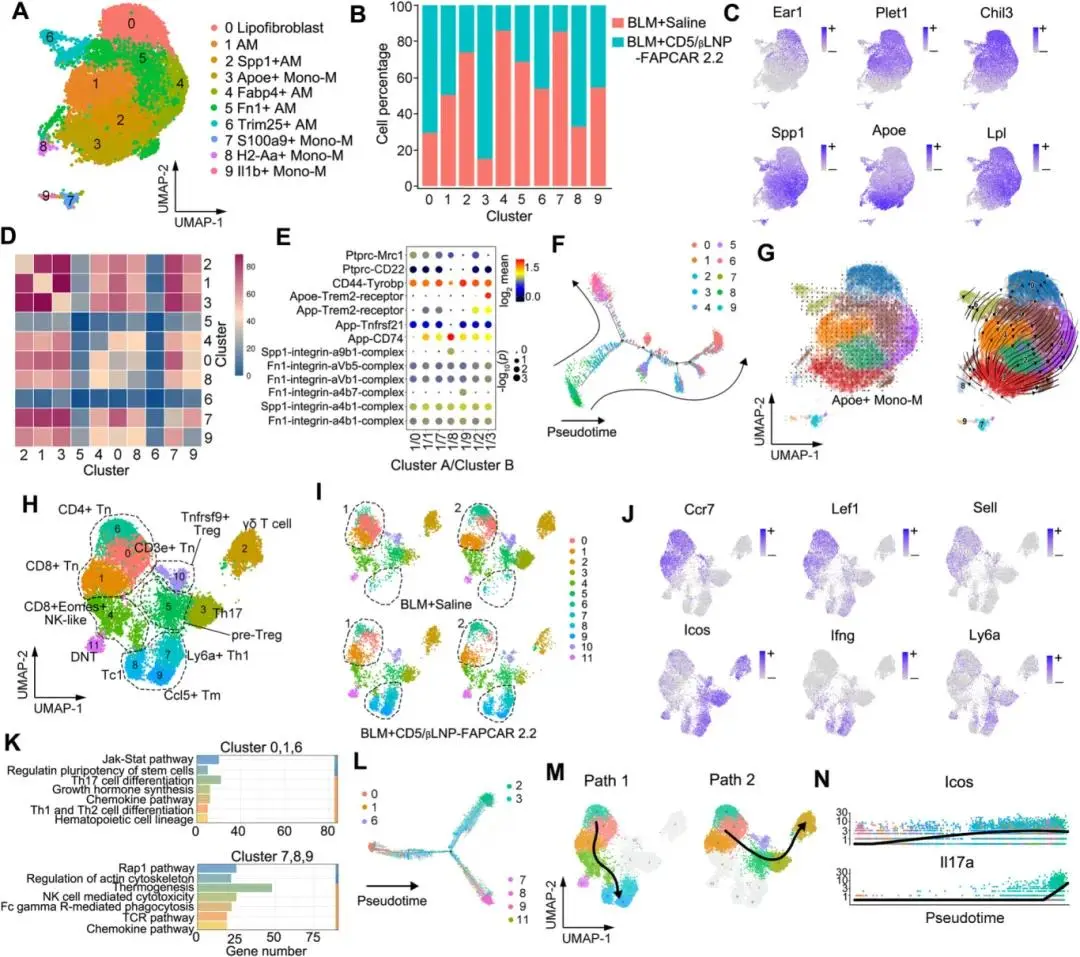
图8. A. 重新聚类的巨噬细胞簇的UMAP图。B. 不同簇中的细胞百分比。 C. 肺泡、纤维化和脂质相关巨噬细胞的标记基因的突出显示。 D. 使用CellphoneDB分析巨噬细胞簇对之间的相互作用。 E. 簇1/3通讯中显著变化的配体-受体对。P值由Bimod测试确定。 F,G. 伪时间(F)和RNA速度(G)分析ApoE+单核细胞的发育速率。 H,I. 重新聚类的T细胞簇的UMAP图:簇注释(H)和两组(BLM+生理盐水与BLM+CD5/βLNP-FAPCAR 2.2)之间的比较(I)。J. 比较BLM+生理盐水和BLM+CD5/βLNP-FAPCAR 2.2组的T细胞簇。K. T细胞簇的KEGG富集分析。L,M. 伪时间(L)和Slingshot(M)分析Tn细胞的分化轨迹。N. T细胞簇中诱导型T细胞共刺激因子(Icos)和白细胞介素-17a(Il17a)的表达水平,随着发育轨迹的增加而增加。(AM:肺泡巨噬细胞;Mono-M:单核细胞衍生的巨噬细胞;Tn:幼稚T细胞;DNT:双阴性T细胞;Tm:记忆T细胞;Tc:细胞毒性T细胞;Th:辅助T细胞;Treg:调节性T细胞)
特发性肺纤维化(IPF)是一种全球范围内广泛存在的严重肺部疾病,但目前可用的临床治疗手段却十分有限。过度活化的成纤维细胞通过异常纤维化堆积来维持炎症与细胞外基质(ECM)硬度之间的动态平衡。由于肺细胞具有再生能力,若能清除这些过度活化的成纤维细胞,肺组织可能具备自我修复功能。该团队研究了瞬时抗纤维化嵌合抗原受体(CAR)T细胞(通过新型脂质纳米颗粒-信使RNA(LNP-mRNA)系统制备)的治疗活性,并探索博来霉素诱导肺纤维化雄性小鼠模型中的肺部再生机制。研究发现,纤维化导致的ECM硬化会损害肺泡上皮细胞的代偿能力。该团队提出的LNP-mRNA疗法通过清除过度活化的成纤维细胞来修复肺纤维化,恢复的ECM环境调控了细胞分布特征。AT2和Pclaf+细胞的可塑性增强促进了AT1细胞群的极化分化。Apoe+巨噬细胞与效应T细胞数量的增加有助于重建肺部免疫功能。因此,LNP-mRNA治疗对纤维化患者的肺结构和功能恢复效果可媲美健康肺组织,为IPF患者提供了潜在的治疗选择。

|
创赛生物 提供高品质的医疗产品和服务 |
联系我们 |
产品中心 |
扫码关注
关注公众号 扫码加客服
|

